

Key Points
The patient reported here, along with collective observations in the literature, suggest that ELANE deletion does not cause neutropenia.
Potential therapeutic genome editing involving knockout of the mutant ELANE allele is therefore not expected to produce neutropenia.
Introduction
Heterozygous mutations in ELANE, encoding neutrophil elastase (NE), cause cyclic neutropenia.1 and are the most common cause of severe congenital neutropenia.2 Numerous patients and mutations are known.3 However, 20 years following discovery, its pathogenesis remains under investigation. One hypothesis is that ELANE mutations result in protein misfolding,4,5 thereby triggering stress responses leading to apoptosis. Another3 posits that mutant polypeptides are mistrafficked yet retain damaging proteolytic activity. Observations that NE inhibitors improve myelopoiesis is interpreted as consistent with either hypothesis.6 Importantly, both hypotheses require that ELANE encodes a mutant polypeptide.
Accordingly, most ELANE mutations represent missense substitutions or in-frame indels, perturbing structures of otherwise intact proteins.3 Nonsense mutations cluster in the terminal exon, where they are predicted to escape nonsense-mediated decay7 and to be expressed as a carboxyl terminally truncated protein.3
Challenging this notion, however, are chain-terminating frameshift mutations located in nonterminal exons,3 expected to undergo nonsense-mediated decay7 with absence of protein production. Similarly, mutations altering the translational start site, at first glance, might also be expected to not produce a protein. In vitro, they initiate translation from internal ATG codons, resulting in amino terminally truncated proteins.8 Whether they do so in vivo, however, has not been determinable. Thus, an open question is whether ELANE loss of function (instead of, or in addition to, gain of function or dominant-negative) mutation contributes to pathogenesis.
To distinguish among hypothesized disease mechanisms, it is therefore important to learn if ELANE whole gene deletions, which are definitively incapable of producing protein, cause neutropenia. The question gains urgency because of proposed therapies using genome editing to delete deleterious alleles encoding gain-of-function mutations.9 Because gene-targeted mouse models do not faithfully recapitulate ELANE-associated phenotypes,10,11 clinical evaluation of patients with ELANE mutations remains among the most powerful approaches for elucidating the molecular pathogenesis of congenital neutropenia.
Heretofore, the hematological consequences of ELANE whole gene deletion have not been studied, for several possible reasons. First, whole gene deletion may be rare, given that ELANE may be subject to unique mutational mechanisms, including as a consequence of its frequent de novo germline mutation and subtelomeric location.12 It is also possible that deletions of ELANE may be particularly deleterious and incompatible with life, or that, conversely, they may be clinically inconsequential and therefore avoid detection.
Here we describe an individual possessing deletion of the entirety of ELANE, occurring in the context of a 37-gene, chromosome 19p terminal deletion.
Case description
The patient is a 21-year-old male of Han Chinese ancestry. His problems include complex congenital heart disease consisting of a double outlet right ventricle and ventricular septal defect surgically corrected in infancy, hypogonadotropic hypogonadism, autism spectrum disorder with intellectual disability, and a diagnosis of Peutz-Jeghers syndrome based on characteristic mucocutaneous hypermelanosis and extensive gastrointestinal polyposis requiring multiple endoscopic and surgical interventions. His parents lack these issues. However, his father has had acromegaly attributed to a pituitary adenoma diagnosed in his third decade of life, and his mother has acute myeloid leukemia with t(8;21) chromosomal translocation, diagnosed at 45 years of age.
The patient has no history of significant recurrent infection, except for aphthous oral ulcers resolving in childhood. Numerous complete blood counts have been performed between the ages of 12 and 21 years (Figure 1). He has not been found to be neutropenic (neutrophil count range, 2.920-12.699 × 109/L), noting that with the sporadic nature of obtaining blood counts, neutropenia occurring cyclically or transiently earlier in childhood cannot be completely excluded. Other complete blood count findings have been unremarkable, except for mild microcytic anemia (hemoglobin concentration range, 8.8-13.2 g/dL; mean cell volume range, 75.7-87.5 fL), attributed to gastrointestinal issues, and mild absolute eosinophilia (range, 0-1.368 × 109/L). Interestingly, eosinophilia is a feature of ELANE-associated neutropenia.13 In this patient, however, eosinophilia, has been thought to coincide with exacerbation of allergies and eczema.
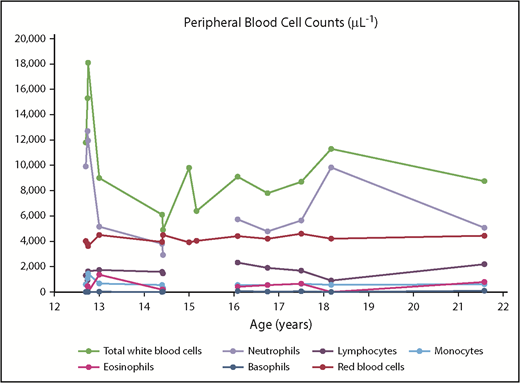
Complete blood counts (peripheral blood), as a function of age (in years). Breaks in lines occur when a complete blood count was performed without corresponding differential cell count.
Complete blood counts (peripheral blood), as a function of age (in years). Breaks in lines occur when a complete blood count was performed without corresponding differential cell count.
Methods
Diagnostic chromosomal microarray analysis performed on the Affymetrix Cytoscan DX array using the patient’s genomic DNA extracted from peripheral blood mononuclear cells revealed an ∼1 Mb heterozygous terminal deletion of the short arm of chromosome 19 (arr[hg19] 19p13.3(260 911-1 319 319) × 1), deleting the following OMIM-listed genes: PLPP2, THEG, C2CD4C, SHC2, MADCAM1, CDC34, GZMM, GSG, HCN2, POLRMT, FGF22, RNF126, FSTL3, PALM, MISP, PTBP1, PLPPR3, AZU1, PRTN3, ELANE, CFD, MED16, KISS1R, ARID3A, GRIN3B, TMEM259, CNN2, ABCA7, ARHGAP45, POLR2E, GPX4, SBNO2, STK11, ATP5F1D, MIDN, CIRBP, and EFNA2. Although neither parent has been similarly tested, the chromosomal deletion is presumptively of de novo origin.
Results and discussion
Ten individuals with similar 19p terminal deletions of variable extent that also include ELANE have been previously reported.14-18 Common features include lentigines and gastrointestinal polyposis consistent with Peutz-Jeghers syndrome (attributed to STK11 deletion), cardiac malformation (attributed possibly to loss of PTBP1), and intellectual disability (ascribed speculatively to deletions of ARID3A and other genes in this region). A unique characteristic of the patient described here appears to be hypogonadotropic hypogonadism, which is ordinarily inherited as an autosomal recessive trait because of biallelic variants in KISS1R,19 suggesting a potential pathogenic mutation contained within the intact chromosome 19.
Of the 10 previously known individuals with a contiguous gene deletion including ELANE, 1 (patient 2 in Peddibhotla et al14 ) was reported to develop neutropenia shortly after birth, albeit in the setting of 33-weeks’ gestational immaturity, hypoglycemia, metabolic acidosis, and necrotizing enterocolitis. The authors speculated that congenital neutropenia in this patient was attributable to ELANE deletion. Further details are lacking, however, including complete blood counts, detection of maternal alloimmune antibodies, bone marrow evaluation, and potential response to treatment with hematopoietic growth factors. Additional description of developmental history to age 4 years does not indicate whether neutropenia and/or infections persisted. Attempts to contact the authors were unsuccessful. Significantly, that patient’s mother and maternal half-sister, who inherited identical chromosome 19p terminal deletions, are not reported to have a history of neutropenia by the ages of 34 and 10 years, respectively,14 suggesting that neutropenia in early infancy was not a consequence of ELANE deletion. Another patient with chromosome 19p terminal deletion including ELANE was described as having immune dysregulation consisting of recurrent otitis and upper respiratory infections, but in that case attributed to low levels of immunoglobulin A and G, responsive to immunoglobulin injections (with no mention of neutropenia).16 Although no blood count information is provided on any of the 10 patients, there is no reference to frequent infections or neutropenia in any of the other reported cases of 19p terminal deletion.
In summary, of 11 patients known to have ELANE whole gene deletion, just 1 was said to have neutropenia. We acknowledge limitations of this present case report, including infrequency of complete blood counts, especially during early childhood, and (clinically justifiable) absence of bone marrow examination, with ELANE expression correspondingly not analyzed. Nevertheless, this patient and the collective observations in the literature suggest that ELANE deletion does not cause neutropenia. Current theories related to gain-of-function mutations leading to unfolding and/or mistrafficking of neutrophil elastase are therefore not excluded. We conclude that, although loss of NE activity may conceivably impair neutrophil function and increase vulnerability to infection,20 potential therapeutic genome editing involving knockout of the mutant allele9 is not expected to produce neutropenia.
Acknowledgments
The authors thank Dawn L. Earl and Amy E. Geddis (Seattle Children’s Hospital) for referring the patient to the Seattle Cancer Care Alliance Hematologic Genetics Malignancy Clinic.
Authorship
Contribution: M.S.H. and M.Y.L. clinically evaluated the patient; S.B.K. consulted on the patient before clinical evaluation; and M.S.H. and S.B.K. wrote the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marshall S. Horwitz, University of Washington School of Medicine, 850 Republican St, Room N435, Seattle, WA 98109; e-mail: horwitz@uw.edu.

