

Key Points
Neutralization of IFN-γ with emapalumab can reverse severe, refractory hemophagocytic lymphohistiocytosis.
Neutralizing IFN-γ did not impair control of multiple viral and other infections in a severely ill patient.

Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an immune-regulatory disorder treated conventionally with corticosteroids and etoposide.1,2 Patients with HLH generally have a poor prognosis and there is considerable morbidity and mortality, largely related to persistent HLH, intercurrent infections, or organ failure.3 Conventional therapy may contribute to these problems due to its myelosuppressive or broadly immunosuppressive effects. Although data are limited, it is generally believed that the prognosis is worse for patients who do not respond to standard treatments.4 Salvage therapy for refractory HLH often involves T-cell–depleting agents, such as alemtuzumab, which produce profound and prolonged immune suppression.5 Animal models have demonstrated a strong causal role for interferon γ (IFN-γ) in the development of HLH,6-9 and data from patients with either primary or secondary HLH have demonstrated elevated levels of IFN-γ or IFN-induced chemokines.8,10-12 These findings have led to the development of emapalumab (NI-0501; Novimmune SA), a fully human anti–IFN-γ monoclonal antibody, which is being tested in a phase 2/3 study as therapy for primary HLH (NCT01818492) and was recently approved by the US Food and Drug Administration. It is hoped that the development of newer targeted therapies such as emapalumab will improve survival in patients with HLH by directly targeting disease-driving pathways and avoiding the myelosuppressive effects of etoposide and the global/persistent immunosuppression of serotherapies.
Case description
We report here a case in which refractory, Epstein-Barr virus (EBV)–associated HLH was successfully treated under an emergency investigational new drug application with the investigational agent emapalumab, despite severe preexisting comorbidities, including multiple life-threatening infections.
Methods
Clinical data are from the Cincinnati Children’s Hospital clinical laboratories. Chemokine levels were measured by Novimmune via MSD, a Meso Scale Discovery assay. Parental consent was obtained for participation in institutional review board–approved research studies, per the Declaration of Helsinki.
Results and discussion
The patient is a boy of southeast Asian descent who presented at 20 months of age with acute EBV infection and essentially all diagnostic features of HLH as defined by the HLH2004 criteria (fever, hepatosplenomegaly, bicytopenia, hypofibrinogenemia, hyperferritinemia, elevated soluble CD25 [sCD25], and marrow hemophagocytosis; natural killer function was not measurable because circulating natural killer cell numbers were too low). At initial presentation, he was noted to have 3.1 million copies of EBV per microgram of DNA in the blood, and cytomegalovirus (CMV) was detected in the urine (but not blood). He was initially treated with etoposide and dexamethasone per the HLH94 protocol and also received rituximab and IV immunoglobulins. Despite this therapy, his clinical status progressively deteriorated. His cytopenias rapidly worsened after initiation of etoposide and his absolute neutrophil count remained below 500 for >5 weeks. After 1 month of therapy, he was transferred to our hospital at which time he had persistence of all HLH features including daily fevers, worsening pancytopenia, and persistent EBV viremia of 3.8 million copies per microgram of DNA.
Shortly after transfer, the patient had severe gastrointestinal bleeding, seizures, and central nervous system hemorrhage, leading to intubation and mechanical ventilation. He also experienced acute renal failure requiring continuous renal replacement therapy, then intermittent hemodialysis. Infectious workup revealed multiple viremias (EBV, CMV, and adenovirus; Figure 1), bacteremia (Escherichia coli), and fungemia (Trichosporon species), all present concurrently. Brain magnetic resonance imaging revealed a large frontoparietal hematoma, later thought to represent a fungal abscess. Notably, CMV was present at very high levels in the blood (18 million IU/mL), whereas adenovirus was detectable at more modest levels (1000-30 000 copies per milliliter in blood). Further standard therapy for HLH was deemed to be futile and salvage therapy with alemtuzumab was considered to be contraindicated due to the presence of severe infections. The patient was not enrolled in the current emapalumab treatment trial (because he met the study exclusion criterion of multiorgan failure), but was treated on an emergency investigational new drug as a last therapeutic resort, though his death was felt to be imminent. Treatment with emapalumab was initiated along with antiviral (ganciclovir, later switched to brincidofovir for adenoviremia), antifungal (liposomal amphotericin), and antibacterial medications. The patient received transfusions of multiple blood products (averaging 5 products per day initially), including daily neutrophil transfusions, for the treatment of fungemia, bleeding, and anemia. Dexamethasone was initially continued, but was then stopped by day 11 due to concern regarding persistent fungemia.
![Figure 1. HLH disease features and viremias after treatment with emapalumab. (A-B) Absolute neutrophil and platelet counts, both heavily supported by transfusion until about day 21. (C-D) Fibrinogen (supported by fresh-frozen plasma transfusion until day 21) and D-dimer levels. (E-F) Alanine aminotransferase (ALT) and direct bilirubin levels. (G-H) The inflammatory markers ferritin and soluble interleukin-2 receptor (sIL2r [sCD25]). Reported ferritin values are limited to 40 000 μg/L by the clinical laboratory. Ferritin values decreased to <500 μg/L by day 180. (I-J) Blood levels of EBV and CMV. Arrows on the x-axis of each graph indicate the start of emapalumab treatment, and initial and final values for each marker are shown.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/1/10.1182_bloodadvances.2018025858/7/m_advances025858f1.png?Expires=1767726835&Signature=eMJA1KVO39UIAkPPA~oK3PJrErdiJhQEj6Xd8KZAZJhOrhTRVn1OZIbRSS5ziSan~rAUDfUC4XL6PuGrWbt0VGN4tTakJ4OdD6wyTNc~ShZxjOh9VcEjDcNnB00EJW6QvJpJ9sqxxMaO4u8O0Ge18kGV5cw1PHVQpYGKsLBjYbftq9~vjOd2GnNiGngCzXig7xtRPNwXcxjta01PBWWWOYDINGD4BvmH-L~1TAnnhIADGlizjdV8jxDn4EmG3m-zIAFnRVJKHmNfzAoVIXJx7dWrCrayT8v-ooKJd7TPzY2HV8mbv5-GHhW21UpQlrZAbHxWGUu41zx09ssqdey10w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HLH disease features and viremias after treatment with emapalumab. (A-B) Absolute neutrophil and platelet counts, both heavily supported by transfusion until about day 21. (C-D) Fibrinogen (supported by fresh-frozen plasma transfusion until day 21) and D-dimer levels. (E-F) Alanine aminotransferase (ALT) and direct bilirubin levels. (G-H) The inflammatory markers ferritin and soluble interleukin-2 receptor (sIL2r [sCD25]). Reported ferritin values are limited to 40 000 μg/L by the clinical laboratory. Ferritin values decreased to <500 μg/L by day 180. (I-J) Blood levels of EBV and CMV. Arrows on the x-axis of each graph indicate the start of emapalumab treatment, and initial and final values for each marker are shown.
HLH disease features and viremias after treatment with emapalumab. (A-B) Absolute neutrophil and platelet counts, both heavily supported by transfusion until about day 21. (C-D) Fibrinogen (supported by fresh-frozen plasma transfusion until day 21) and D-dimer levels. (E-F) Alanine aminotransferase (ALT) and direct bilirubin levels. (G-H) The inflammatory markers ferritin and soluble interleukin-2 receptor (sIL2r [sCD25]). Reported ferritin values are limited to 40 000 μg/L by the clinical laboratory. Ferritin values decreased to <500 μg/L by day 180. (I-J) Blood levels of EBV and CMV. Arrows on the x-axis of each graph indicate the start of emapalumab treatment, and initial and final values for each marker are shown.
Treatment of HLH with emapalumab alone resulted in resolution of all clinical symptoms and normalization of clinical laboratory parameters (Figure 1). Within hours of receiving the first dose of emapalumab, the patient’s fever resolved. Approximately 1 week after initiating emapalumab, serum ferritin began to fall rapidly; sCD25 fell with slightly slower kinetics. Within 2 weeks of starting emapalumab, gastrointestinal bleeding resolved, fluid and neurologic status improved, and the patient was extubated. Transfusion support was weaned off by day 21 and neutrophil and platelet counts progressively improved, reaching normal levels within 2 months. Hepatic dysfunction and coagulopathy resolved within a month. Renal function recovered (glomerular filtration rate increased to >60 mL/min) over 3 months. The patient received emapalumab for 14 weeks. Treatment was well tolerated with no clinically significant adverse reactions noted.
Remarkably, during blockade of IFN-γ with emapalumab, all infections resolved with supportive antimicrobial medications and cessation of etoposide and dexamethasone. Bacteremia did not persist and fungemia resolved by day 14 of treatment. EBV fell rapidly starting 1 week after emapalumab and became undetectable by day 160 (Figure 1). CMV viremia resolved in ∼3 weeks and adenoviremia resolved by 7 weeks. By the end of therapy, all HLH-related symptoms had resolved, all infections resolved (brain hematoma/abscess eventually resolved ex vacuo), the patient had made a complete neurologic recovery, and he was able to stop dialysis. Hematopoietic stem cell transplant was not pursued because the patient had no suitable donors and genetic workup did not reveal HLH-associated gene mutations. The patient was discharged from the hospital after 174 days, and he has been off of all therapy for >36 months with no evidence of unusual infections, recurrent EBV, or HLH. He is developing normally and is in excellent health.
Three features of this case are remarkable. First, despite prolonged prior immunosuppressive therapy, the patient had extremely elevated CXCL9 and CXCL10 levels (Figure 2). These chemokines are specifically produced in response to IFN-γ and such levels suggest severely elevated in vivo IFN-γ bioactivity.13 Second, these chemokines promptly decreased upon the initiation of emapalumab and fell to normal or near normal levels within about a week. Together with the clinical response, these data indicate that emapalumab was able to neutralize IFN-γ and that such monotherapy (albeit with dexamethasone for the first 10 days) effectively halted severe and refractory HLH. Although animal models have predicted this effect, this is the first clear demonstration of such therapeutic potential in humans. Third, despite IFN-γ neutralization, this patient was able to recover from multiple infections that would ordinarily be considered unsurvivable. Although treatment with emapalumab allowed for cessation of conventional immunosuppressive therapy, which was likely impeding infection control, our findings call into question the role that IFN-γ plays in antiviral resistance in humans. Interferons, both type I (α/β) and type II (γ), are thought to play important roles in viral resistance. However, our patient was able to clear persistent EBV (and other infections) while on emapalumab, even though supportive antivirals are of little or no benefit for EBV infection. Paradoxically, our findings suggest that IFN-γ may have been contributing to immune paralysis in this patient. Experimental models have demonstrated immune paralysis in response to type I interferons only,14,15 though IFN-γ is well described as an inducer of various immune-regulatory pathways.16,17 This patient’s course suggests that IFN-γ blockade is a rational and attractive strategy for targeted treatment of HLH, and agents such as emapalumab will teach us much about the biology of IFN-γ in humans.
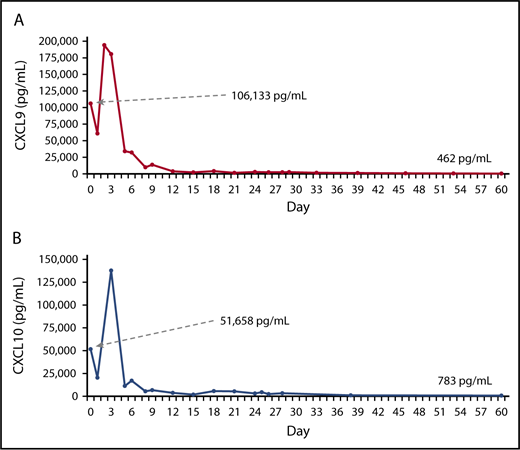
IFN-γ–associated chemokines after treatment with emapalumab. The levels of CXCL9 (A) and CXCL10 (B), 2 chemokines induced by IFN-γ, after initiation of therapy with emapalumab. Baseline and final levels for each biomarker are indicated.
IFN-γ–associated chemokines after treatment with emapalumab. The levels of CXCL9 (A) and CXCL10 (B), 2 chemokines induced by IFN-γ, after initiation of therapy with emapalumab. Baseline and final levels for each biomarker are indicated.
Acknowledgments
The authors thank Didier Halimi and Innotio for assistance with editing and figure preparation.
Authorship
Contribution: D.T.L. and M.B.J. wrote the report; Q.B. and C.d.M. assisted in gathering data and/or editing the manuscript; and C.d.M. additionally provided chemokine data.
Conflict-of-interest disclosure: C.d.M. is an employee of Novimmune SA. M.B.J. is a consultant for, and serves on a scientific advisory board for, Novimmune. The remaining authors declare no competing financial interests.
Correspondence: Michael B. Jordan, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, ML7038, Cincinnati, OH 45229; e-mail: michael.jordan@cchmc.org.

