

Key Points
B microti infection induces a robust adaptive immune response in WT mice with coordinated development of GC reaction and Ag-specific Abs.
Sickle mice develop low parasitemia, but elicit a strong adaptive immune response despite a disorganized splenic architecture preinfection.

Abstract
The intraerythrocytic parasite Babesia microti is the number 1 cause of transfusion-transmitted infection and can induce serious, often life-threatening complications in immunocompromised individuals including transfusion-dependent patients with sickle cell disease (SCD). Despite the existence of strong long-lasting immunological protection against a second infection in mouse models, little is known about the cell types or the kinetics of protective adaptive immunity mounted following Babesia infection, especially in infection-prone SCD that are thought to have an impaired immune system. Here, we show, using a mouse B microti infection model, that infected wild-type (WT) mice mount a very strong adaptive immune response, characterized by (1) coordinated induction of a robust germinal center (GC) reaction; (2) development of follicular helper T (TFH) cells that comprise ∼30% of splenic CD4+ T cells at peak expansion by 10 days postinfection; and (3) high levels of effector T-cell cytokines, including interleukin 21 and interferon γ, with an increase in the secretion of antigen (Ag)-specific antibodies (Abs). Strikingly, the Townes SCD mouse model had significantly lower levels of parasitemia. Despite a highly disorganized splenic architecture before infection, these mice elicited a surprisingly robust adaptive immune response (including comparable levels of GC B cells, TFH cells, and effector cytokines as control and sickle trait mice), but higher immunoglobulin G responses against 2 Babesia-specific proteins, which may contain potential immunogenic epitopes. Together, these studies establish the robust emergence of adaptive immunity to Babesia even in immunologically compromised SCD mice. Identification of potentially immunogenic epitopes has implications to identify long-term carriers, and aid Ag-specific vaccine development.
Introduction
Human babesiosis is caused by the protozoan parasite Babesia microti, which invades and multiplies within human red blood cells (RBCs). Infections vary greatly in their presentation depending on age and immune competency of the host. Severe symptoms present in neonates,1-4 or in older adults, possibly due to depressed cellular immunity5-7 and in the immunocompromised of any age, particularly asplenic individuals.8-14 Mild disease caused by B microti usually presents with intermittent fever with general malaise and weakness.15 In patients who were hospitalized with severe B microti infection, death occurred in ∼10% of cases.16,17
B microti can be transmitted through blood transfusions, and remains the foremost infectious risk to the US blood supply18-20 for which a Food and Drug Administration (FDA)–licensed test is still unavailable for donation screening, with tests under investigational new drug not performing optimally according to available data.21 Most blood centers still rely on a blood donor questionnaire to self-identify any previous history of babesiosis.20
Mechanisms underlying Babesia infection and host immune response remain poorly understood. There are no studies to date on the nature of the immune response in individuals infected with B microti, with most available data coming from the parasitized mouse models. The primary B microti infection, which is generally self-limiting and nonlethal, generates a strong protection against a subsequent infection,22 and can be passively transferred to recipient mice by the transfer of splenocytes.23,24
Macrophages in innate immunity are critical for the clearance of Babesia-infected RBCs as well as for driving resistance to infection.25 In addition, T-cell–mediated immunity has been shown to protect against B microti infection,22,26-31 although the exact mechanism of the protection against a subsequent infection remains unknown.22,26 In addition, B cells appear to be involved in the clearance of Babesia, although in 1 study, it was deemed that they had a minimal role in the control of acute parasemitia.26 Furthermore, various immunoregulatory cells have been implicated in the immune response to Babesia.24,32 However, there are no data regarding the contribution of adaptive immune cell compartments during B microti infection. For example, little is known about the kinetics of the induction of germinal center (GC) reaction, which is critical for the development of B-cell responses and the generation of plasma cells secreting high-affinity antibodies and memory B cells.33-37 Nor is much known about the role of T follicular helper (TFH) cells, which promote humoral immunity by providing help to B cells and facilitating the selection of plasma and memory cells33,38,39 in the adaptive response to B microti infection and parasite clearance. Understanding the kinetics of adaptive immunity against Babesia infection can help determine the pathogenesis of babesiosis and account for persistent infection.26
Patients with sickle cell disease (SCD) require transfusions, which dramatically increases their risk of transfusion-transmitted infections40 ; however, little is known about B microti pathogenesis in human SCD that results in disease exacerbation.13 An altered baseline immune function in mice SCD confounded by impaired splenic function may underlie exacerbated infections,41 as demonstrated by either a hyporesponsive immune response or a hyperresponsive immune response, depending on the types of antigens to which they have been exposed.42 For example, whereas vaccination against both Streptococcus pneumoniae and the influenza A virus produces low titers of antigen-specific immunoglobulin G (IgG) and IgM43-45 and defects in viral clearance likely ascribed to decreased levels of interferon γ (IFNγ) following influenza challenge in SCD mice,46 experimental asthma induces an exaggerated immune response, as shown by increased antigen-specific IgE production.47-49 The observation that sickle trait, heterozygous for hemoglobin S (HbS), confers protection against malarial infection, whereas homozygous HbS is associated with increased morbidity and mortality in both humans and mice,50-55 is suggestive of impaired immunity in SCD. However, there exists limited knowledge on immune functions in the context of SCD, especially adaptive immunity.
In this study, we investigated the kinetics of adaptive immune response to acute Babesia infection in mice to determine the extent to which adaptive immunity contributes to babesiosis and whether an impaired immune response in SCD would result in lethal outcomes following B microti infection.
Material and methods
Mice
C57BL/6J (000664), transgenic SCD HbSS-Townes mice, and HbAS-Townes mice, were purchased from The Jackson Laboratory (Bar Harbor, ME). The HbSS-Townes mice (referred to as SS; homozygous for βS) were created by knocking-in human α and βS globins into the locus of murine α and β globins, leading to the expression of only human globins.56 HbAS-Townes mice (referred to as AS) have human α globin but are heterozygous for human βA and βS globins. The HbAA-Townes control mice (referred to as AA) were created by replacing βS with βA and obtained by breeding the HbAS-Townes mice. Both male and female mice, 9 to 12 weeks old, were used for this study; animals were housed in microisolator cages in a special pathogen-free facility. All animal studies were approved by the New York Blood Center’s Animal Care and Use Committee.
Parasite infections and detection
The B microti (Franca) Reichenowi Peabody strain was obtained from ATCC (Manassas, VA). Mice were infected via intraperitoneal injection with 107 to 108B microti–parasitized RBCs. Parasitemia was determined using standard methods for collecting a drop of anticoagulated blood from the tail vein and using this blood to perform Giemsa staining and flow cytometry.
Light microscopy.
Blood smears were fixed with methanol and stained with 20% Giemsa (Sigma-Aldrich, St. Louis, MO) for the morphological analysis of parasites and for evaluation of parasitemia. A minimum of 2000 cells was counted for parasitemia measurement or scanned for assessment of changes in morphology using a Nikon Eclipse E 600 microscope.
Flow cytometry.
The size of the in vivo B microti–infected RBC population was measured by flow cytometry, by performing a novel dual-color staining protocol, developed in our laboratory, modifying a protocol previously designed for Babesia divergens.57 Mouse erythrocytes were identified by allophycocyanin (APC) rat anti-mouse TER-119 (BD Pharmingen). Infected RBCs were identified by staining parasite DNA using Hoescht 33342 (2′-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]-2, 5′-bi-1H-benzimidazole trihydrochloride trihydrate) (Thermo Fisher Scientific). As all RBCs lack a nucleus, RBCs with a positive signal for DNA represent infected host cells bearing parasites. To avoid inaccurate parasitemia determinations because of the presence of RNA in reticulocytes, the preferential binding of Hoechst to A-T regions of DNA was exploited. APC–TER-119 antibody was used at 0.005 μM and Hoechst at 0.1 μM final concentrations at a concentration of 1 × 107 cells per milliliter. Samples were analyzed on a LSR Fortessa SORP analyzer (BD Biosciences), equipped with a 355-nm UV laser for Hoechst detection (361/486 nm) and a 640-nm red laser for APC–TER-119 detection (650/60 nm BP). The forward scatter threshold was set on 5000, and 10 000 total events were acquired at low flow rate. FACSDiva software (version 6.2; BD Biosciences) was used for data analysis. All parameters were processed using log scaling.
Flow cytometry and antibodies for immune studies
Various tissues including spleens and lymph nodes along with peripheral blood were collected from mice on the indicated days following B microti infection, and the spleens were weighed. Single-cell suspensions were prepared by gently meshing the spleens and lymph nodes in RPMI 1640 medium containing 10% fetal bovine serum. The cells were incubated with anti-CD16/32 (FRC-4G8) at 4°C for 15 minutes in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA). For flow cytometry, lymphocytes (1 × 106 cells) were incubated with fluorescence-labeled or biotinylated monoclonal antibodies (mAbs) followed by fluorescent streptavidin, and analyzed as previously described.58 APC-labeled anti-Foxp3 (FJK-16s) mAbs, phycoerythrin (PE)/Cy7-labeled anti-IgM (II/41) mAb, PE-labeled anti-IgD (11-26c) mAb, fluorescein isothiocyanate (FITC)-labeled anti–programmed death 1 (PD-1; J43) mAb, anti-CD16/CD32 (FRC-4G8) mAb, peridinin chlorophyll protein (PerCP)/Cy5.5-labeled anti-IFNγ (XMG1.2) mAb, and APC-labeled anti–interleukin 4 (IL-4; 11B11) mAb were purchased from Thermo Fisher.
Alexa F700 or Pacific Blue–labeled anti-CD4 (RM4-5), PerCP/Cy5.5-labeled anti-CD278 (inducible costimulator [ICOS]; C398.4A), Alexa F700–labeled anti-CD44 (IM7), Pacific Blue–labeled anti-CD45R/B220 (RA3-6B2), and PerCP/Cy5.5-labeled streptavidin were purchased from BioLegend. Biotinylated anti-CD95 (Fas; Jo2), FITC-labeled anti–T- and B-cell activation antigen (GL7), APC-labeled anti-CD138 (281-2) mAb, biotinylated anti-CXCR5 (2G8) mAb, PE or PE-CF594–labeled anti-Bcl-6 (K112-91) mAb, and PE-Cy7–labeled streptavidin were purchased from BD Biosciences. Recombinant mouse IL-21R Fc chimera protein was purchased from R&D Systems. R-Phycoerythrin AffiniPure F(ab′)2 fragment goat anti-human IgG, Fcγ fragment-specific, antibody was purchased from Jackson ImmunoResearch Laboratories. For the staining of regulatory T cells (Tregs), APC anti-mouse Foxp3 (Thermo Fisher) was intracellularly stained according to the manufacturer’s instructions. For the intracellular staining of IL-21, splenocytes were stimulated with phorbol myristate acetate (50 ng/mL) and ionomycin (1 μM; EMD) for 4 hours in the presence of protein transporter inhibitor brefeldin A (1 μg/mL; BD Biosciences). After surface staining, the cells were fixed with Cytofix/Cytoperm buffer (BD Biosciences) for 30 minutes and incubated with recombinant mouse IL-21R Fc chimera protein for 30 minutes on ice, followed by the staining with a PE-labeled affinity purified F(ab′)2 fragment of goat anti-human Fcγ antibody. Flow cytometric data were analyzed with FlowJo software (TreeStar).
Immunofluorescence microscopy
Immunofluorescence staining was performed as reported previously.58 Briefly, spleens of mice were frozen after embedding in OCT compound (Tissue-Tek; Sakura). Frozen blocks were cut into 6 μm in thickness with a microtome at −20°C and dried >2 hours at room temperature. Sections were fixed in cold acetone for 15 minutes and kept at −80°C. Sections were blocked with 3% BSA (serum-free; Jackson ImmunoResearch Laboratories) in PBS for 20 minutes. Cryosections were incubated with primary antibodies, PE-conjugated anti-IgD (11-26c.2a; Thermo Fisher), APC-conjugated anti-CD3ε (145-2C11; Thermo Fisher), and FITC-conjugated peanut agglutinin (PNA; Vector Laboratories) for staining of GCs. Images were captured using an inverted LSM 780 laser-scanning confocal microscope (Zeiss).
Immunohistochemistry and H&E staining
Histology and immunohistochemistry slide preparation and interpretation were performed at the Laboratory of Comparative Pathology of Memorial Sloan Kettering Cancer Center, Weill Cornell Medicine, and The Rockefeller University. Tissues were fixed in 4% paraformaldehyde, processed routinely in alcohol and xylene, embedded in paraffin, sectioned at 5-μm thickness, and stained with hematoxylin-eosin (H&E). Immunohistochemistry (IHC) staining for B220 and CD3 was performed on a Leica Bond RX automated stainer (Leica Biosystems, Buffalo Grove, IL). Following heat-induced epitope retrieval at pH 6.0 (B220) and 9.0 (CD3), the primary antibody against B220 (BD Pharmigen 550286) and CD3 (Abcam ab16669) was applied at a concentration of 1/200 and 1/100, respectively, followed by application of a polymer detection system including secondary antibodies (Novocastra Bond Polymer Refine Detection; Leica Biosystems), according to the manufacturer’s instructions. The chromogen was 3,3 diaminobenzidine tetrachloride, and sections were counterstained with hematoxylin. Interpretation of H&E and IHC slides was performed by a board-certified veterinary pathologist.
Luminex immunoassay
The concentrations of plasma antibody class/subclass in naive and B microti–infected mice were measured using the Milliplex Antibody Isotyping 7-Plex Mouse ProcartaPlex Panel 2 Immunoassay according to the manufacturer’s instructions, as previously described.59 Briefly, the diluted mouse plasma (1/40 000) was incubated with the antibody magnetic beads (IgG1, IgG2a, IgG2b, IgG3, IgA, IgE, IgM) followed by antibody detection beads. The concentration of 7 antibodies was calculated by plotting the expected concentration of the standards against the mean fluorescence intensity generated by each standard and the concentrations adjusted to account for the dilution.
ELISA for parasite-specific immunoglobulins
B microti crude extract preparation and ELISA.
Following B microti infection, when the parasitemia was ∼60%, mice were scarified and RBCs used to prepare the crude lysate by controlled lysis of the parasite-infected erythrocytes with 0.1% saponin in 0.06 M NaCl, ultrasonication in the presence of enzyme inhibitors, centrifugation at 4°C, and filtration on a 0.2-mm pore-size filter (Millex). Protein concentrations were determined, and extracts were frozen at −80°C in working aliquots.
Total B microti antigen (100 μL of 10 μg/mL) in 50 mM sodium bicarbonate, pH 9.4, was used to coat enzyme-linked immunosorbent assay (ELISA) plates overnight at 4°C. Then, 200 μL of blocking buffer (2% BSA in PBS with 0.05% Tween-20) was added to each well and the plates incubated for 1 hour at 37°C. Serum samples were serially diluted from 1/100 to 1/100 000 in a dilution buffer (1% BSA in PBS with 0.05% Tween-20) and 100 μL of diluted serum was distributed into each well (each sample tested in triplicate). Polyclonal anti-mouse IgG (1/5000; GE Healthcare) or anti-mouse IgAGM (1/2000; KPL) conjugated to peroxidase was added. Bound peroxidase was detected with sureblue substrate (KPL) 20 minutes (100 μL) and the reaction was stopped by addition of 2 M H2SO4 (50 μL per well). Between each incubation phase, the ELISA plates were washed extensively with PBS–0.05% Tween-20. The optical density (OD) at 450 nm was read in a Bio-Rad microplate reader.
Library ELISA.
Biotinylated B microti recombinant proteins produced using a mammalian expression system were immobilized in individual wells of streptavidin-coated microtiter plates as described in detail previously.60 Briefly, 26 B microti genes that are predicted to encode surface-displayed and secreted proteins were expressed by transient transfection in suspension-grown HEK293E. 6-His–tagged proteins were purified from spent tissue culture supernatant. Protein levels were quantified by ELISA after immobilizing dilutions of the biotinylated CD4-tagged protein in individual wells of streptavidin-coated microtiter plates and using the mouse anti-rat CD4 monoclonal antibody (OX68) as a detection antibody and an anti-mouse alkaline phosphatase conjugate as a secondary antibody. To quantify elicited mouse antibody titers, proteins were immobilized directly on streptavidin-coated plates as above (using the mouse anti-rat CD4 monoclonal antibody). Sera was diluted 1/2000 in 2% (wt/vol) BSA-PBS, and total mouse IgG binding was detected using anti-mouse alkaline phosphatase–conjugated secondary antibody.
Statistical analysis
Statistical analysis was performed with Graphpad Prism 7 and statistical significance was analyzed using unpaired 2-tailed Student t tests. Statistical details of experiments are described in the figure legends. P < .05 was considered significant. *P < .05, **P < .01, ***P < .001, and ****P < .0001.
Results
Kinetics of B microti infection in C57BL/6J mice
Intraperitoneal B microti infection with 107 to 108 parasitized RBCs in multiple experiments resulted in robust infection in C57BL/6J mice as detected by flow cytometry as well as Giemsa-stained blood smears (supplemental Figure 1A-E). However, there was 100% survival in the mice following infection (data not shown). Consistent with previous data,26 the levels of B microti parasitemia exponentially increased between day 3 and day 7, reaching maximal levels at day 7 postinfection with as high as 48% ± 1% of erythrocytes being infected, followed by parasite clearance (<1%) by day 21 postinfection (Figure 1A). IV parasite infections produced similar kinetics of parasitemia, albeit with faster initial establishment of infection (data not shown). Erythrocyte numbers and hemoglobin content were significantly reduced (from 9.6 ± 0.2 × 106/μL and 13.2 ± 0.7 g/dL to 4.1 ± 0.3 × 106/μL and 5.7 ± 0.5 g/dL, respectively) by day 7 postinfection and began to recover from day 21 onwards upon clearance of parasites and returning to an almost normal range (8.7 ± 0.2 × 106/μL and 12.9 ± 0.3 g/dL, respectively) at day 38 postinfection (Figure 1B-C). In contrast, reticulocyte counts steadily increased reaching 60% by day 10 postinfection but were on a decline by day 21 postinfection (Figure 1D). Infection induced splenomegaly as previously reported61,62 (supplemental Figure 2A) with an approximately ninefold increase in spleen weights (Figure 1E), and 4.5-fold increase in total splenocyte numbers by day 14 postinfection (Figure 1F), which interestingly lagged behind peak parasitemia on day 7.
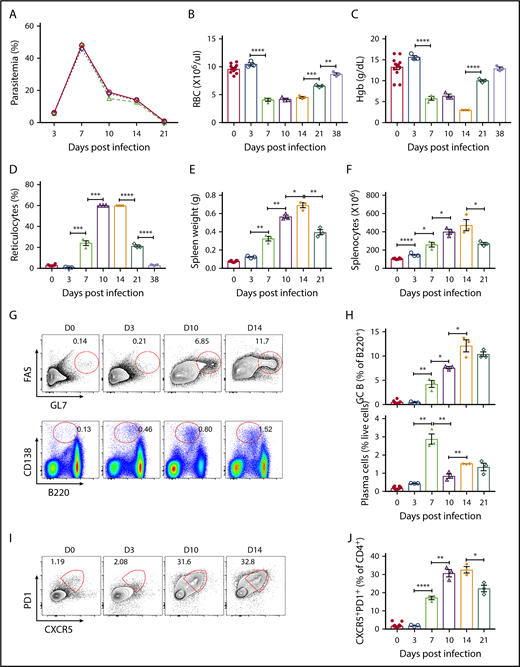
Kinetics of adaptive immune response following B microti infection in C57BL/6 mice. Groups of C57BL/6 mice were infected with 1 × 107 to 1 × 108B microti–parasitized RBCs intraperitoneally and a representative experiment is shown. (A) The frequency of peripheral blood erythrocytes infected with B microti in total number of RBCs over time. (B-D) Peripheral blood on the indicated days following B microti infection were analyzed using an Advia 120 hematology system for absolute numbers of RBCs (B), hemoglobin (Hgb) content (C), and reticulocyte percentages (D) (n = 3-10). (E-F) The spleen weights (E) and the number of splenocytes (F) before and after B microti infection (n = 3-12). (G) Representative flow cytometry (FACS) plots for GL7 and FAS gated on B220+ cells for the identification of splenic GC B cells (top panel) or B220 and CD138 for the identification of plasma cells (bottom panel) before infection (D0) and after infection on days 3, 10, and 14 (D3, D10, and D14). (H) Quantification of GC B cells (top panel) and plasma cells (bottom panel) (n = 3-10). (I-J) Flow cytometric analysis of TFH cells in C57BL6 mice. (I) Representative FACS plots for CXCR5 and PD1 expression gated on splenic CD4+ T cells. (J) Quantification of TFH cells (CD4+CXCR5hiPD1hi) (n = 3-10). Each symbol represents data from an individual mouse. Error bars indicate the mean ± standard error of the mean (SEM). *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired 2-tailed Student t test.
Kinetics of adaptive immune response following B microti infection in C57BL/6 mice. Groups of C57BL/6 mice were infected with 1 × 107 to 1 × 108B microti–parasitized RBCs intraperitoneally and a representative experiment is shown. (A) The frequency of peripheral blood erythrocytes infected with B microti in total number of RBCs over time. (B-D) Peripheral blood on the indicated days following B microti infection were analyzed using an Advia 120 hematology system for absolute numbers of RBCs (B), hemoglobin (Hgb) content (C), and reticulocyte percentages (D) (n = 3-10). (E-F) The spleen weights (E) and the number of splenocytes (F) before and after B microti infection (n = 3-12). (G) Representative flow cytometry (FACS) plots for GL7 and FAS gated on B220+ cells for the identification of splenic GC B cells (top panel) or B220 and CD138 for the identification of plasma cells (bottom panel) before infection (D0) and after infection on days 3, 10, and 14 (D3, D10, and D14). (H) Quantification of GC B cells (top panel) and plasma cells (bottom panel) (n = 3-10). (I-J) Flow cytometric analysis of TFH cells in C57BL6 mice. (I) Representative FACS plots for CXCR5 and PD1 expression gated on splenic CD4+ T cells. (J) Quantification of TFH cells (CD4+CXCR5hiPD1hi) (n = 3-10). Each symbol represents data from an individual mouse. Error bars indicate the mean ± standard error of the mean (SEM). *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired 2-tailed Student t test.
B microti infection induces a robust GC reaction, and a significant expansion of TFH cells
To uncover a potential protection mechanism against B microti infection in the spleen, we investigated the adaptive immune response in the spleens of B microti–infected C57BL/6J mice. During GC formation, GC B cells downregulate the surface expression of IgD, whereas they increase the surface expression of GL7 and Fas.35,36,63,64 By contrast, plasma cells downregulate the surface expression of B220, whereas they express high levels of CD138.65-68 GC B cells (GL7+Fas+B220+) were strongly induced upon B microti infection beginning on day 7 at 4.2% ± 0.7% as compared with <1.5% before infection and reaching 12.1% ± 1.2% by day 14 after which point there was a trend toward a decline, although, surprisingly, the levels remained high (10.3% ± 0.5%) even at day 21 postinfection when parasitemia is resolved (Figure 1G-H top panels), consistent with a robust B-cell immune response that continued to increase even after peak parasitemia on day 7.
Interestingly, we observed very high percentages of splenic plasma cells (CD138+B220low) ∼2.8% ± 0.3% on day 7 postinfection, which is most likely induced primarily by T-independent immune responses. The percentage of plasma cells was maintained at high >1% until day 21 postinfection (Figure 1G-H bottom panels).
We also examined whether the TFH compartment, identified as CXCR5+PD1+CD4+ by flow cytometry,58 is impacted during B microti infection. We detected a significant development of TFH cells upon parasite infection, with 31% ± 2% of CD4+ T cells becoming TFH cells on day 10 and remaining high (33% ± 2%) through day 14 postinfection (Figure 1G,I, as compared with <3% before infection). The kinetics of TFH cell development paralleled that of GC B cells, suggesting a well-coordinated induction of adaptive immune response against B microti infection (Figure 1D-G).
T-cell effector cytokines and plasma immunoglobulins following B microti infection
To evaluate T-cell effector function during B microti infection, we determined the levels of cytokines over the course of infection, focusing on IL21, a critical cytokine for the GC formation, and TFH and B-cell differentiation and immunoglobulin class switching38,69-75 and IFNγ, which is implicated in the pathophysiological function of TFH cells and protection against Babesia32,76 as well as immunoglobulin switching.74,77 We found a significant induction of IL21 as well as IFNγ with ∼8% and 7% of CD4+ T cells, respectively, in splenic CD44+ effector CD4+ T cells as early as day 5 postinfection (Figure 2A).
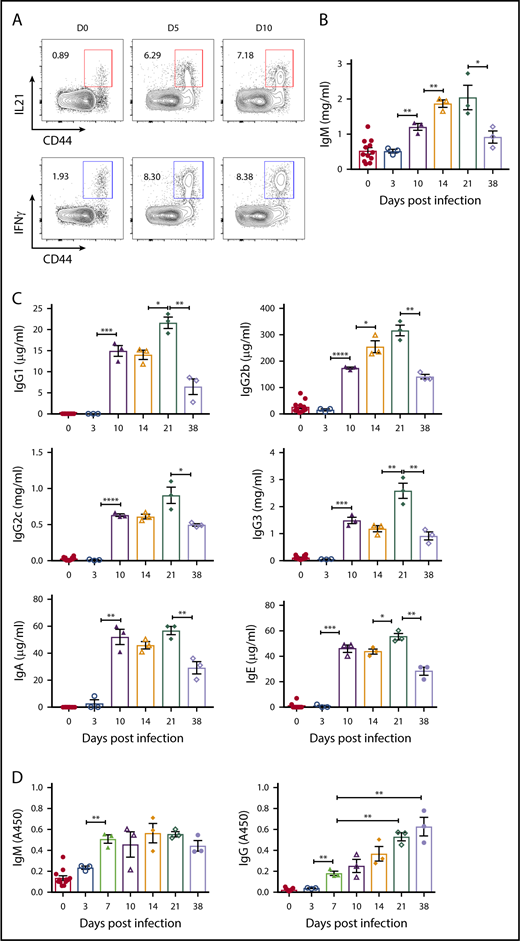
CD4+T cells expressing cytokines and the kinetics of plasma immunoglobulin levels following B microti infection in C57BL6 mice. (A) Representative FACS plots for CD44 and IL21 (top) or IFNγ (bottom) expression gated on CD4+ T cells in the spleens from mice before (D0) and after B microti infection on days 5 and 10. (B-C) Plasma immunoglobulin levels of IgM (B), IgG1, IgG2b, IgG2c, IgG3, IgA, and IgE (C) on days indicated following Babesia infection. The concentrations of each immunoglobulin isotype were determined by Luminex assay with appropriate immunoglobulin standards. (D) The kinetics of B microti–specific antibody responses in C57BL/6 mice following infection. Plasma was collected from mice on the days indicated and the levels of IgM and IgG antibodies were determined by ELISA using crude cell lysate of RBCs infected with B microti. Each symbol represents data from an individual mouse. Error bars indicate the mean ± SEM. *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired 2-tailed Student t test.
CD4+T cells expressing cytokines and the kinetics of plasma immunoglobulin levels following B microti infection in C57BL6 mice. (A) Representative FACS plots for CD44 and IL21 (top) or IFNγ (bottom) expression gated on CD4+ T cells in the spleens from mice before (D0) and after B microti infection on days 5 and 10. (B-C) Plasma immunoglobulin levels of IgM (B), IgG1, IgG2b, IgG2c, IgG3, IgA, and IgE (C) on days indicated following Babesia infection. The concentrations of each immunoglobulin isotype were determined by Luminex assay with appropriate immunoglobulin standards. (D) The kinetics of B microti–specific antibody responses in C57BL/6 mice following infection. Plasma was collected from mice on the days indicated and the levels of IgM and IgG antibodies were determined by ELISA using crude cell lysate of RBCs infected with B microti. Each symbol represents data from an individual mouse. Error bars indicate the mean ± SEM. *P < .05; **P < .01; ***P < .001; ****P < .0001 by unpaired 2-tailed Student t test.
We next measured the titers of class-switched total immunoglobulins by Luminex assay to examine the humoral immune responses during Babesia infection. Consistent with expression of effector cytokines involved in class switching, we found a significant increase in the plasma levels of all isotypes tested including IgM (≅1.2 mg/mL), IgG1 (≅15 μg/mL), IgG2b (≅170 μg/mL), IgG2c (≅0.6 mg/mL), IgG3 (≅1.5 mg/mL), IgA (≅50 μg/mL), and IgE (≅45 μg/mL) antibodies by day 10, followed by a decrease on day 38 postinfection (Figure 2B-C). In addition, using whole-cell extracts from parasitized RBCs, we detected a gradual increase of Babesia-specific IgM antibody levels beginning at day 3 (OD ≅ 0.2) until day 38 (OD ≅ 0.5), when the levels started declining (Figure 2D left panel). Interestingly, the levels of Babesia-specific IgG antibodies were continually increasing at day 38 (OD ≅ 0.7) postinfection, the last point we measured (Figure 2D right panel), suggestive of an active ongoing adaptive immune response.20
Townes mice, a murine model of human SCD, have low levels of parasitemia following B microti infection
We next evaluated the adaptive immune response of the Townes SCD mouse model56 following infection with B microti. Berkeley SCD mice have been reported to have severely disrupted splenic architecture, leading to impaired immunity against pathogens.41 As controls, we used AA mice that express 2 normal (A) copies, or trait AS mice expressing 1 normal (A) and 1 sickle (S) human Hb gene, respectively. Based on multiple experiments, we found that AA and AS mice exhibited similar levels of parasitemia, but surprisingly SS mice consistently presented with at least fourfold to fivefold lower levels of parasitemia (see example Figure 3A, approximating 20% parasitemia at peak levels in AA and AS mice but only 4% to 5% in SS mice). All parasite forms were visible in the 3 strains and looked identical morphologically (supplemental Figure 1E). All mice, irrespective of genotype, were eventually able to clear their infections.

Kinetics of adaptive B-cell immune response following B microti infection in Townes SS and control mice. Groups of Townes SS mice along with control AA and AS mice were infected with 1 × 107 to 1 × 108B microti–parasitized RBCs intraperitoneally and a representative experiment is shown. (A) The frequency of peripheral blood erythrocytes infected with B microti in total number of RBCs. (B) The spleen weights of AA, AS, SS mice before and after infection. (C) Representative FACS plots for GL7 and FAS gated on B220+ cells for the identification of GC B cells in the spleens from AA, AS, and SS mice. (D) Quantification of GC B cells in the spleens from AA, AS, and SS mice on days indicated before and following infection. (E) Representative FACS plots for B220 and CD138 for the identification of plasma cells. (F) Quantification of plasma cells in the spleens from AA, AS, and SS mice. Each symbol represents data from an individual mouse. Error bars indicate the mean ± SEM. **P < .01; ***P < .001; ****P < .0001 by unpaired 2-tailed Student t test. ns, not significant.
Kinetics of adaptive B-cell immune response following B microti infection in Townes SS and control mice. Groups of Townes SS mice along with control AA and AS mice were infected with 1 × 107 to 1 × 108B microti–parasitized RBCs intraperitoneally and a representative experiment is shown. (A) The frequency of peripheral blood erythrocytes infected with B microti in total number of RBCs. (B) The spleen weights of AA, AS, SS mice before and after infection. (C) Representative FACS plots for GL7 and FAS gated on B220+ cells for the identification of GC B cells in the spleens from AA, AS, and SS mice. (D) Quantification of GC B cells in the spleens from AA, AS, and SS mice on days indicated before and following infection. (E) Representative FACS plots for B220 and CD138 for the identification of plasma cells. (F) Quantification of plasma cells in the spleens from AA, AS, and SS mice. Each symbol represents data from an individual mouse. Error bars indicate the mean ± SEM. **P < .01; ***P < .001; ****P < .0001 by unpaired 2-tailed Student t test. ns, not significant.
Spleen weights of AA and AS mice gradually increased ≅ threefold following B microti infection, whereas that of SS mice, despite the high basal levels (≅ sixfold increased spleen weights compared with those of AA and AS), were still marginally enlarged after infection (Figure 3B; supplemental Figure 2B).
B microti infection induces a vigorous GC reaction in Townes mice despite low levels of parasitemia
Given the low levels of parasitemia compared with AA and AS mice, as well as an eventual resolution of B microti infection even in SS mice, we examined the adaptive immunity in the spleens of SS mice. As expected from higher frequencies of cells in myeloid lineages in naive SS mice, we observed low levels of overall frequencies of B and T cells in the spleens of uninfected SS mice (supplemental Figure 3A-B). Although the frequencies of B cells remained low over the course of Babesia infection, the frequencies of T cells increased to a level similar to those of AA and AS mice at day 10 postinfection (supplemental Figure 3A-B). Despite lower levels of parasitemia (Figure 3A), SS mice surprisingly mounted a very strong adaptive immune response following B microti infection, as shown by high percentages of GC B cells on days 5 and 10 (>12%) postinfection as compared with <1.5% before infection, and more importantly similar to levels induced in AA or AS mice (Figure 3C-D; P > .1). A similar pattern of GC reaction in all 3 strains of mice was detected even on day 17 (supplemental Figure 4A) when parasitemia had almost cleared (supplemental Figure 2B-C) after which point a decrease in GC B-cell percentages was finally apparent (supplemental Figure 4A day 21). Interestingly, we detected a similar trend of immune responses in the lymph nodes from all 3 strains of mice postinfection, suggesting that SS mice induce overall similar levels of GC reaction following B microti infection (supplemental Figure 4B). As with GC B cells, we observed high percentages of splenic plasma cells (CD138+B220low) in SS mice at days 5 and 10 postinfection (1.7% ± 0.2% and 1.5% ± 0.2%, respectively) similar to those of AA or AS mice (Figure 3E-F) with a detectable decline in percentages on day 21 postinfection (data not shown).
To gain a better insight into the enhanced adaptive immune response in the spleens of SS mice, we next evaluated the size and distribution of splenic GC B cells by immunofluorescence imaging (Figure 4). As expected, spleens from naive AA or SS mice have very small to no GCs (Figure 4 PNA shown in green). Notably, spleens from naive SS mice showed very small follicles identified by IgD staining (Figure 4A shown in red). Furthermore, we detected indistinct boundaries between the B-cell zone and T-cell zone identified by IgD (in red) and CD3 staining (in blue, white arrows), respectively, in the spleens from naive SS mice compared with those of AA (Figure 4A) or AS mice (data not shown). Remarkably, following Babesia infection, spleens from SS mice formed distinct, well-organized GCs similar to those of AA mice (Figure 4B).
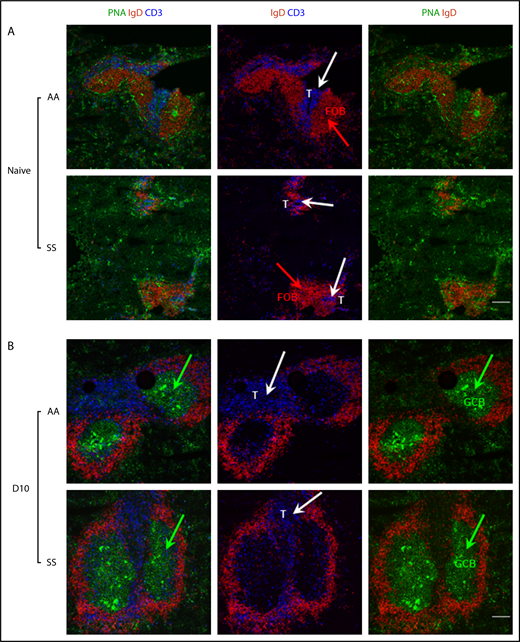
Immunofluorescence analysis of splenic T- and B-cell zones of Townes SS and control mice before and following B microti infection. Representative immunofluorescence images of splenic sections from uninfected (A) and B microti–infected (B) Townes SS and control AA mice (n = 2-3 mice) stained for PNA (a GC marker, using FITC-conjugated PNA, in green), IgD (a B-cell marker, using PE-conjugated anti-IgD, red), and CD3 (a T-cell marker, using APC-conjugated anti-CD3e, blue). Although there is a clear T-cell (blue)/B-cell (red) boundary in the spleens from naive AA mice, T cells are located independently of B-cell follicle, without a clear boundary in the spleens from naive SS mice. GCs marked by PNA in green are formed similarly in both AA and SS mice following Babesia infection. Green arrows indicate clusters of PNA+ GC B cells (GCB); white arrows indicate CD3+ T cells. Scale bars, 200 μm. FOB, follicular B cell.
Immunofluorescence analysis of splenic T- and B-cell zones of Townes SS and control mice before and following B microti infection. Representative immunofluorescence images of splenic sections from uninfected (A) and B microti–infected (B) Townes SS and control AA mice (n = 2-3 mice) stained for PNA (a GC marker, using FITC-conjugated PNA, in green), IgD (a B-cell marker, using PE-conjugated anti-IgD, red), and CD3 (a T-cell marker, using APC-conjugated anti-CD3e, blue). Although there is a clear T-cell (blue)/B-cell (red) boundary in the spleens from naive AA mice, T cells are located independently of B-cell follicle, without a clear boundary in the spleens from naive SS mice. GCs marked by PNA in green are formed similarly in both AA and SS mice following Babesia infection. Green arrows indicate clusters of PNA+ GC B cells (GCB); white arrows indicate CD3+ T cells. Scale bars, 200 μm. FOB, follicular B cell.
To evaluate whether spleens from SS mice have a unique splenic architecture following B microti infection enabling them to form such well-developed GCs, we performed IHC staining for B220 and CD3. Similar to the Berkeley SCD mouse model,42 SS Townes mice showed overtly disorganized splenic architecture in the naive state (Figure 5A). Specifically, we detected big follicles with a distinct T/B boundary in the spleens from AA and C57BL/6 mice (Figure 5A). However, consistent with immunofluorescence staining with IgD, spleens from naive SS mice have very small, indistinct follicles identified by B220 (blue arrows), which is found in overlapping regions with T cells identified by CD3 (red arrows) (Figure 5A). CD3+ T cells were dispersed around the periarteriolar lymphatic sheathes, making an indistinct T/B boundary in SS mice, whereas they were densely populated around the well-developed periarteriolar lymphatic sheathes in AA (Figure 5A) and AS mice (data not shown). Interestingly, we observed marked changes in splenic architecture following B microti infection, but, surprisingly, the overall organization of the white pulp of the spleens was relatively comparable in all 3 strains postinfection with significantly enlarged follicles in all mice at day 10 (Figure 5B). This was accompanied by the development of distinct GCs that were relatively well-organized and comparable in size in all 3 strains (Figures 4B and 5B, and data not shown) with loosening of a clear T-cell zone boundary in both AA and SS mice.
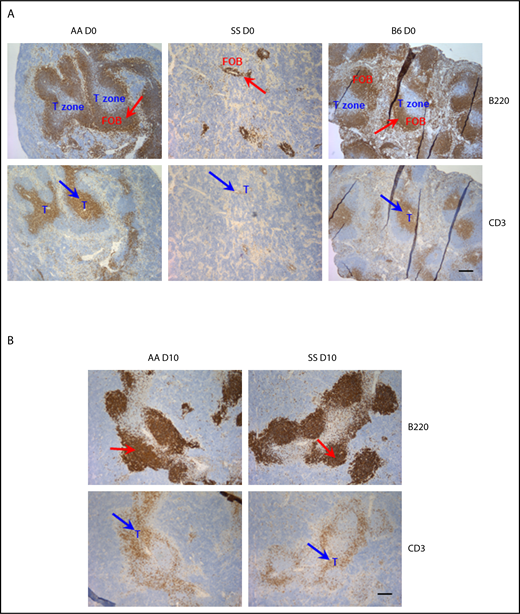
Analysis of splenic follicles of Townes SS and control mice before and following B microti infection. Representative immunohistochemical staining of splenic follicles in Townes SS and control AA and C57BL/6 mice before (A) or after (B, on day 10) B microti infection. Splenic sections were stained for B220 (and detected using horseradish peroxidase (HRP)–linker antibody conjugate with DAP as substrate, brown) and CD3 (and detected using HRP-linker antibody conjugate with DAP as substrate, brown) (n = 2-3 mice). T cells and follicular B cells (FOB) are localized in nonoverlapping areas in the spleens from naive, uninfected C57BL/6, and AA mice. Spleens from naive, uninfected SS mice shows very small indistinct follicles. Following Babesia infection (B), large GCs are formed in the spleens from both AA and SS mice. Red arrows indicate B220+ B-cell follicles; blue arrows indicate CD3+ T cells. T-cell zone is marked in blue. Scale bars, 200 μm.
Analysis of splenic follicles of Townes SS and control mice before and following B microti infection. Representative immunohistochemical staining of splenic follicles in Townes SS and control AA and C57BL/6 mice before (A) or after (B, on day 10) B microti infection. Splenic sections were stained for B220 (and detected using horseradish peroxidase (HRP)–linker antibody conjugate with DAP as substrate, brown) and CD3 (and detected using HRP-linker antibody conjugate with DAP as substrate, brown) (n = 2-3 mice). T cells and follicular B cells (FOB) are localized in nonoverlapping areas in the spleens from naive, uninfected C57BL/6, and AA mice. Spleens from naive, uninfected SS mice shows very small indistinct follicles. Following Babesia infection (B), large GCs are formed in the spleens from both AA and SS mice. Red arrows indicate B220+ B-cell follicles; blue arrows indicate CD3+ T cells. T-cell zone is marked in blue. Scale bars, 200 μm.
TFH cell development, T effector cytokines, and Tregs in B microti–infected Townes mice
In agreement with a strong GC reaction, we detected high frequencies of CXCR5+PD1+CD4+ TFH cells in all 3 strains (on day 5 with 9% ± 2%, 11% ± 2%, and 12% ± 1% in AA, AS, and SS, respectively, as compared with <6% before infection in all strains), which increased to 39% ± 4%, 40% ± 3%, and 38% ± 1% in AA, AS, and SS, respectively, on day 10 postinfection (Figure 6A-B). Interestingly, we observed slightly lower expression of ICOS, a critical costimulatory molecule in the generation of class-switched antibodies,78 in TFH cells at the beginning of the immune response in SS mice compared with that of AA and AS mice, suggesting a potential qualitative difference in SS TFH cells (supplemental Figure 3C). However, this difference was eliminated by day 10 postinfection (supplemental Figure 3C). Even when parasitemia began to clear on day 17 (supplemental Figure 2C), TFH cell frequencies were high and remained comparable in AA, AS, and SS mice (supplemental Figure 4C). As with GC B cells, overall TFH cell frequencies began to decrease after day 17 postinfection (supplemental Figure 4C). We also found a similar increase in the frequency of TFH cells in the lymph nodes in all 3 strains (supplemental Figure 4D).
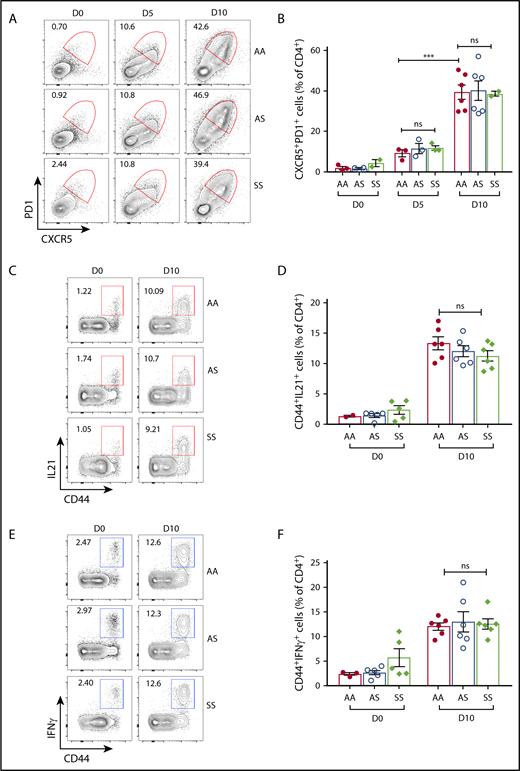
Kinetics of T-cell immune response following B microti infection in Townes SS and control mice. (A-B) Flow cytometric analysis of TFH cells in Townes SS and control AA and AS mice following B microti infection. (A) Representative FACS plots for CXCR5 and PD1 expression gated on splenic CD4+ T cells. (B) Quantification of TFH cells (CD4+CXCR5hiPD1hi) (n = 2-6). Combined data from 3 independent experiments. (C) Representative FACS plots for CD44 and IL21 expression gated on splenic CD4+ T cells. (D) Quantification of IL21+ cells in the spleens from AA, AS, and SS mice. (E) Representative FACS plots for CD44 and IFNγ expression gated on splenic CD4+ T cells. (F) Quantification of IFNγ+ cells gated in the spleens from AA, AS, and SS mice. Each symbol represents data from an individual mouse. Error bars indicate the mean ± SEM. ***P < .001; by unpaired 2-tailed Student t test.
Kinetics of T-cell immune response following B microti infection in Townes SS and control mice. (A-B) Flow cytometric analysis of TFH cells in Townes SS and control AA and AS mice following B microti infection. (A) Representative FACS plots for CXCR5 and PD1 expression gated on splenic CD4+ T cells. (B) Quantification of TFH cells (CD4+CXCR5hiPD1hi) (n = 2-6). Combined data from 3 independent experiments. (C) Representative FACS plots for CD44 and IL21 expression gated on splenic CD4+ T cells. (D) Quantification of IL21+ cells in the spleens from AA, AS, and SS mice. (E) Representative FACS plots for CD44 and IFNγ expression gated on splenic CD4+ T cells. (F) Quantification of IFNγ+ cells gated in the spleens from AA, AS, and SS mice. Each symbol represents data from an individual mouse. Error bars indicate the mean ± SEM. ***P < .001; by unpaired 2-tailed Student t test.
In line with a strong adaptive immune response following B microti infection, we detected a robust increase of IFNγ+ T cells in the spleens from all 3 strains of mice with 13% ± 1%,16% ± 3%, 18% ± 1% of CD44+ memory CD4+ T cells in AA, AS, and SS, respectively, on day 5 postinfection and comparable frequencies at day 10 (Figure 6C-D), suggesting a similar effector function of T cells in SS mice. We also found a similar trend in the expression of IL21 in all 3 mouse strains (Figure 6E-F). These data suggest that T effector cells in SS mice have a similar effector function in controlling the acute B microti infection during the earlier phase of Babesia infection.
We hypothesized that the Tregs in SS mice may be altered causing defects in regulating the function of T effector cells, which may result in the expansion of T effector cells including TFH cells, leading to an efficient adaptive immune response. However, we did not detect any differences among the 3 different strains of mice (supplemental Figure 4E), indicating that Treg cells in infected SS mice are at least quantitatively similar to infected AA and AS mice.
Plasma immunoglobulins and Babesia-specific antibody response in Townes mice
To evaluate whether there are altered immunoglobulins in the plasma of infected SS mice, we first measured total immunoglobulin levels in the plasma of infected mice over the course of Babesia infection using the Luminex assay. We observed similar levels of all of the immunoglobulins measured following Babesia infection (IgG1 [≅12 μg/mL], IgG2b [≅300 μg/mL], IgG2c [≅0.8 mg/mL], IgA [≅20 μg/mL], and IgE [≅15 μg/mL]) except for the levels of IgM on day 10 postinfection, which showed a twofold decrease in SS mice (data not shown), consistent with the deficiency of marginal zone B cells, known for IgM production, in SS mice and humans.79,80 These data indicate that overall levels of total switched immunoglobulins are very similar in all 3 strains of B microti–infected mice.
We next considered the possibility that there may be a difference in the Babesia-specific antibody repertoire enabling them to respond differently against infection. We have previously identified immunoreactive extracellular parasite proteins from B microti against humans and mice.60 Using 26 such proteins (supplemental Table 1), we observed 9 proteins that induced a strong antibody response against B microti infection in C57BL/6 mice (supplemental Figure 4). A similar pattern of reactivity was detected in AA, AS, and SS mice toward the later part of the infection, around day 17 postinfection (Figure 7). However, Bm32 (a putative copper transport protein) and, even more so, Bm34 (a sporozoite microneme protein), respectively,81 were reproducibly found to elicit a significantly higher antibody response in SS mice as compared with AA or AS mice (1.3- to 1.4-fold and fourfold to ninefold, respectively; Figure 7B).
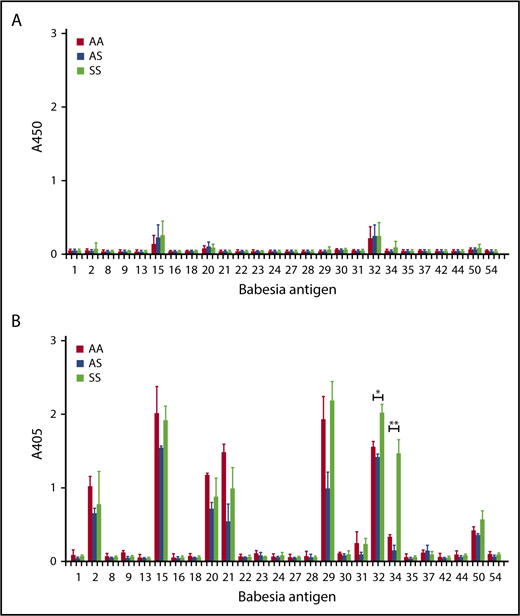
Analysis of B microti–specific IgG responses. Plasma was collected from mice on day10 (A) and day 17 (B) in Townes (AA, AS, SS) mice following B microti infection and the levels of Babesia-specific IgG antibody were determined by ELISA using recombinant B microti proteins as listed in supplemental Table 1 (n = 2-3). Error bars indicate the mean ± SEM. *P < .05; **P < .01; by unpaired 2-tailed Student t test.
Analysis of B microti–specific IgG responses. Plasma was collected from mice on day10 (A) and day 17 (B) in Townes (AA, AS, SS) mice following B microti infection and the levels of Babesia-specific IgG antibody were determined by ELISA using recombinant B microti proteins as listed in supplemental Table 1 (n = 2-3). Error bars indicate the mean ± SEM. *P < .05; **P < .01; by unpaired 2-tailed Student t test.
Discussion
In this study, we have investigated the immune response against B microti infection with a special focus on a quantitative understanding of the kinetics of adaptive immune responses to infection using murine models. We found that Babesia infection in C57BL/6 mice induced a very strong adaptive immune response, characterized by a robust GC reaction, and significant expansion of TFH cells with as much as one-third of CD4+ T cells becoming TFH cells. By comparison, <10% of CD4+ T cells developed into TFH cells following a conventional T-dependent antigen immunization with alum-precipitated 4-hydroxy-3-nitrophenylacetyl-chicken gamma globulin (data not shown). The kinetics of TFH cell development were similar to that of GC B cells, suggesting a well-coordinated induction of adaptive immune response against B microti infection. Although it is not known whether these GCs harbor antigen-specific cells, the GC/TFH response along with production of effector T-cell cytokines IL-21 and IFNγ, led to an increase in the secretion of antigen-specific antibodies over time. Interestingly, we detected a delay in the kinetics of the GC/TFH cell expansion compared with those of parasitemia. For example, GC/TFH cells reached the peak on day 14 postinfection following the peak of parasitemia on day 7 postinfection in C57BL/6 mice. A similarly strong GC/TFH cell response, but yet delayed, is also observed after infection with Plasmodium chabaudi chabaudi in mice.82 The significance of this exaggerated adaptive immune response against Babesia and Plasmodium infection, however, is currently not known. Our finding that B microti infection induces GC B cells supports a role for B cells in control of Babesia infection and parasite clearance, and is consistent with the reports of persistent babesiosis in patients treated with B-cell–depleting rituximab.83,84 It is also likely that TFH cells are equally important for controlling B microti given that a crucial role of TFH cells in the clearance of RBC parasites has been demonstrated in a lethal Plasmodium berghei ANKA infection, where compromised TFH cell development resulted in an impaired GC reaction leading to severe malaria.85
A striking finding of this study was that Babesia induced a very robust adaptive immune response in SCD mice, despite very low levels of parasitemia. The overall levels of adaptive immune response, such as the proportion of GC B cells and TFH cells, were very similar in control (AA and AS) and SS mice despite a significant difference in the levels of parasitemia. This is surprising considering that uninfected naive SS mice have massively disrupted splenic architecture, which may inhibit the obligatory interactions between B and T cells during GC reactions. We therefore had expected to detect very low to no immune response against B microti infection, causing the mice to develop severe babesiosis and even death. Instead, we found that despite a disorganized splenic architecture, the collaboration between T and B cells to form GCs surprisingly does occur following B microti infection in SCD mice. The formation of GCs and plasma cells independently of the discrete T-/B-cell boundary has been reported in malarial infection, suggesting that B cells can form fully functional GCs in collaboration with the follicular dendritic cells in the absence of distinct follicles.82 Remarkably, the marginal zone B cells that facilitate antigen transport to follicular dendritic cells is also significantly reduced in naive SS mice (data not shown) and yet the immune response to Babesia is apparently intact. Interestingly, the selective loss of IgM antibody secretion due to a loss of IgMhiIgDlowCD27+ “IgM memory B cells” residing in the marginal zone was also observed in patients with SCD,79,80 suggesting that marginal zone B cells may also be absent in human SCD. The discrepancies of the kinetics between the levels of parasitemia and the degrees of adaptive immunity in SS mice imply that there may be an unknown immune pathway in the spleens of SS mice, which enable them to induce an efficient adaptive immune response.
The observed heightened immune response against Babesia infection in SCD can be explained by at least 2 premises. First, SCD mice may have a stronger immune system against Babesia infection than AA and AS mice. However, we did not find increased percentages of the immune cells, including GC or TFH cells. Instead, we have found that 2 of the Babesia-specific epitopes, Bm32 and, even more robustly, Bm34, induced significantly higher Babesia-specific IgG responses toward the later part of the infection in SS mice as compared with AA or AS mice. Bm32 (BMN1-20) is a 59-kDa member of the immunodominant BMN family and has been putatively identified as a copper transport protein analogous to transporter proteins seen in Plasmodium and Theileria.86-88 Bm34 has been identified as a sporozoite microneme protein.81 It is a 96-kDa protein located in the apically situated micronemes of the parasite. Interestingly, Bm34 contains a membrane attack complex domain, which is known to be responsible for pore formation in target membranes in a number of systems and could be used by the parasite to porate the RBC membrane to facilitate invasion or egress. However, no characterization of either Bm32 or Bm34 protein has been carried out yet and the effects of these epitopes in response to Babesia infection, especially in adaptive immunity, are currently not known. If these epitopes are critical factors among various stages of Babesia infection, including the initial attachment, penetration, and internalization into the RBC, then the increased immune response in SCD mice against these 2 proteins would lead to inhibition of the expansion of parasitemia despite no apparent increase in the other parameters of adaptive immunity, including GC B cells and TFH cells relative to AA and AS mice. As such, a detailed study of these molecules relating to their role in pathogenesis may provide clues as to why the SCD mice respond so strongly to these antigens and whether this response plays a role in the resistance to hosting the high parasite loads seen in the sickle trait (AS) and wild-type (AA or C57BL/6) mice. Our findings of the differences of antibody repertoire will be very useful in future studies, such as in the search for immunoreactive antigens of Babesia, which can improve a diagnosis of human long-term carriers and the development of antigen-specific vaccines.
A second explanation for hyperresponsiveness to Babesia infection in SS mice is that these mice may have a very sensitive immune system, enabling them to respond strongly to even very low levels of parasitemia via an enhanced recognition of parasites. This can be explained if SCD mice are resistant to the Babesia infection but respond very strongly to low levels of parasitemia. SCD mice have a substantial increase in the levels of reticulocytes, immature RBCs, relative to mature RBCs, ∼40% to 80% compared with 4% in AA mice.56 It has been shown that Babesia multiply better in mature RBCs than in reticulocytes, shown by the differences in the percentages of intracellular parasites. For example, <10% of reticulocytes contain intracellular parasites at any point of the infection whereas >70% of mature RBCs contain parasites on day 7 following Babesia infection.26 As such, the overall infection rate can be reduced in SCD, relative to AA or AS mice. If SCD mice are equipped with a very sensitive immune recognition system for detecting Babesia, they can induce similar levels of GC/TFH cells in SS mice in response to low levels of parasitemia. Possible mechanisms include an increase in the sensitivity of TCR to antigen recognition or an increase in the antigen-presenting function. This is a feasible possibility because SCD mice are in a chronic inflammatory state, which can serve as the underlying mechanisms to sensitize the inflammation. These data warrant future studies on the potential importance of a memory response to Babesia. In particular, it would be valuable to see whether SCD mice respond better in the subsequent infection because they have a different repertoire of antibodies in the primary infection.
In summary, we report in this study a thorough and quantitative kinetics of adaptive immune response to Babesia infection. Our unexpected finding that SCD mice mount an equally robust adaptive immunity despite low parasitemia underscores the importance of examining both the fate of B microti and the immunological consequences of parasite infection in individuals with SCD to establish whether a similar hyperimmune response against the parasite occurs in humans too. Patients with SCD require transfusions, with some on chronic transfusion therapy, placing them at greater risk of acquiring transfusion-transmitted infections like babesiosis as currently there is no mandate to screen blood for this blood-borne pathogen. As such, these individuals, if transfused from an infected donor, would be exposed to a larger infectious dose compared with a tick bite. Furthermore, chronically transfused patients would be at higher risk of reinfection via transfusion. The outcome of these infections, whether one of immune protection mediated by the first infection or a more deleterious pathological sequel, is required to be studied to establish effective treatments for these patients.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank services provided by the Laboratory of Comparative Pathology of Memorial Sloan Kettering Cancer Center, Weill Cornell Medicine, and The Rockefeller University for animal tissue sections and H&E staining.
This work was supported by Wellcome Trust grant 206194 (G.J.W.) and National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL140625 (C.A.L.) and R01HL130139 (K.Y.).
Authorship
Contribution: W.Y. planned, performed, and analyzed the immunological experiments while W.B. performed the experiments and assisted with the analysis; M.R. performed most of the parasite infections, analysis of parasitemias, and ELISAs for plasma antibodies; Y.L. assisted with infections, blood draws, and whole-mount and immunofluorescence studies and analysis; M.S., H.Z., J.R.C.-S., V.R., and A.M. assisted with experimental procedures and analysis; C.M.E. and G.J.W. provided the B microti expression library; A.M. and X.A. provided input in experimental design in addition to data analysis; K.Y. and C.A.L. were responsible for experimental design, manuscript writing, and project supervisions; W.Y. wrote the manuscript; and K.Y. and C.A.L. edited the manuscript with consultation and contribution from all coauthors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Karina Yazdanbakhsh, New York Blood Center, 310 East 67th St, New York, NY 10065; e-mail: kyazdanbakhsh@nybc.org; and Cheryl A. Lobo, New York Blood Center, 310 East 67th St, New York, NY 10065; e-mail: clobo@nybc.org.

