

Key Points
Rapid initiation of eculizumab mitigates progression of carfilzomib-induced aHUS.
Development of carfilzomib-induced aHUS may be associated with heterozygous CFHR3-CFHR1 deletion.
Introduction
Atypical hemolytic uremic syndrome (aHUS) results from uninhibited continuous activation of the alternative complement pathway and is characterized by acute kidney injury, thrombocytopenia, and microangiopathic hemolytic anemia. Without treatment, most patients with aHUS develop end-stage renal disease. Since the introduction of eculizumab, it is now possible to mitigate disease progression1 by blocking the terminal complement pathway (Figure 1). Eculizumab therapy, given initially as monotherapy (900 mg/wk) or concurrently with apheresis (600 mg per session), is typically initiated after a normal ADAMTS13 activity level rules out thrombotic thrombocytopenic purpura and is continued indefinitely as maintenance therapy. Notably, earlier initiation of eculizumab is associated with greater renal recovery,2 and case reports demonstrate the efficacy of eculizumab treatment specifically in carfilzomib-induced aHUS.3
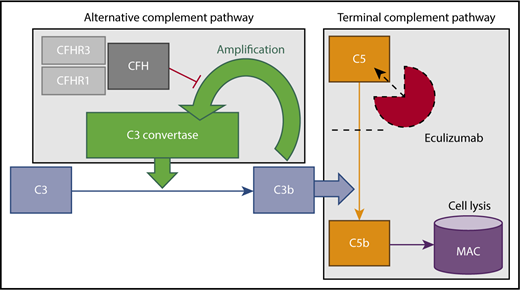
Alternative and terminal complement pathways. Complement factor H (CFH), along with its related proteins (CFHRs), inhibits amplification of C3b. Eculizumab binds C5, preventing conversion to C5b and subsequent formation of the membrane attack complex (MAC), which effects cell lysis and mediates renal epithelial damage.
Alternative and terminal complement pathways. Complement factor H (CFH), along with its related proteins (CFHRs), inhibits amplification of C3b. Eculizumab binds C5, preventing conversion to C5b and subsequent formation of the membrane attack complex (MAC), which effects cell lysis and mediates renal epithelial damage.
Carfilzomib, an irreversible proteasome inhibitor used in relapsed or refractory multiple myeloma, is a known trigger of aHUS. Although a proteasome inhibitor class effect has been proposed as a cause of thrombotic microangiopathies, carfilzomib has been uniquely linked to the development of aHUS.3-5 We present 3 cases of carfilzomib-induced aHUS, a potential mechanism of disease, and considerations for treatment with eculizumab.
Case description
Case report 1
Patient 1 is a 68-year-old African American woman with a history of plasma cell leukemia status postinduction with lenalidomide/bortezomib/dexamethasone and autologous hematopoietic stem cell transplant and subsequently on carfilzomib/lenalidomide maintenance therapy. Her total cumulative dose (TCD) of carfilzomib was 464 mg.
She was admitted on cycle 3, day 5 with 2 days of nausea/vomiting, diarrhea, and oliguria. Initial laboratory tests demonstrated acute kidney injury, with creatinine = 9.09 mg/dL (baseline ∼1.4) and hemolytic anemia, with hemoglobin (Hgb) = 8.7 g/dL, platelets = 4 × 109/L, haptoglobin < 20 mg/dL, and lactate dehydrogenase (LDH) = 1443 U/L. Peripheral blood smear (PBS) contained schistocytes. Direct antiglobulin test (DAT) and stool bacterial polymerase chain reaction were negative. She progressed to anuria, necessitating regular hemodialysis (HD) starting on hospital day 3. She did not receive eculizumab treatment. After 2 weeks, her platelet count began to spontaneously uptrend (Figure 2A). She continued to require HD and has shown no evidence of renal recovery after 2 years.
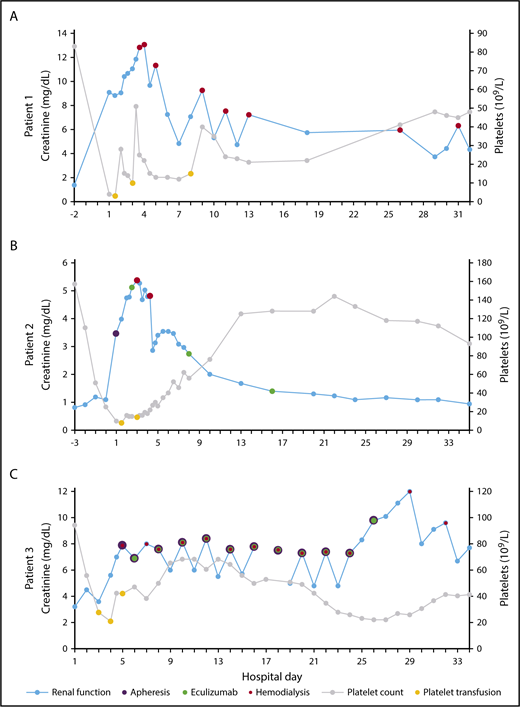
Creatinine and platelet count vs hospital day. HD (red), apheresis (purple), and eculizumab (green) are plotted on creatinine (blue) and platelet transfusions (yellow) are plotted on platelet count (gray) for patient 1 (A), patient 2 (B), and patient 3 (C).
Creatinine and platelet count vs hospital day. HD (red), apheresis (purple), and eculizumab (green) are plotted on creatinine (blue) and platelet transfusions (yellow) are plotted on platelet count (gray) for patient 1 (A), patient 2 (B), and patient 3 (C).
Case report 2
Patient 2 is a 59-year-old white male with type 2 diabetes and multiple myeloma status postinduction with lenalidomide/carfilzomib/dexamethasone and autologous stem cell transplant with very good partial response on carfilzomib and lenalidomide maintenance therapy. His TCD of carfilzomib was 1826 mg.
He was admitted on cycle 4, day 17 with 1 day of fever, myalgia, and generalized weakness. Initial laboratory tests showed acute kidney injury and thrombocytopenia, with creatinine = 1.19 mg/dL (baseline ∼0.8) and platelets = 51 × 109/L (baseline ∼110). He was treated with empiric antibiotics for 2 days, but no infectious source was identified. He was discharged home.
The following day he developed diarrhea, cola-colored urine, and oliguria. He was readmitted with acute renal failure, anemia, and thrombocytopenia. Initial laboratory tests showed creatinine = 3.46 mg/dL, platelets = 8 × 109/L, and Hgb = 9.8 g/dL. Additional laboratory tests were consistent with hemolytic anemia, with total bilirubin = 2.0 mg/dL, haptoglobin < 20 mg/dL, and LDH = 1297 U/L. PBS contained schistocytes. DAT was negative. Emergent apheresis was performed overnight. On hospital day 2, apheresis was discontinued, and treatment was begun with eculizumab 900 mg.
On hospital day 3, his platelet count began to spontaneous uptrend. Urine output steadily improved; however, on hospital days 3 and 4, he required HD for volume overload. Starting on hospital day 6, his creatinine began to spontaneously downtrend (Figure 2B). ADAMTS13 activity level returned at >100%. Eculizumab was continued at 900 mg/wk for 3 total doses. His renal function improved to baseline after 1 month and has remained stable over the past year.
Case report 3
Patient 3 is a 66-year-old white male with plasma cell leukemia who began treatment with cyclophosphamide/carfilzomib. His TCD of carfilzomib was 329 mg.
On cycle 2, day 2 he was admitted with hypotension and generalized weakness in the setting of several days of diarrhea. Initial laboratory tests showed anemia and acute kidney injury, with Hgb = 8.8 g/dL (baseline ∼10) and creatinine = 3.2 mg/dL (baseline ∼1.1). He developed anuria and rapidly worsening thrombocytopenia, with a platelet nadir of 19 × 109/L (hospital day 4). Additional laboratory values were consistent with hemolytic anemia, with total bilirubin = 1.3 mg/dL and LDH = 3960 U/L. PBS contained schistocytes. DAT and stool bacterial polymerase chain reaction were negative. On hospital day 5, he began apheresis and regular HD. ADAMTS13 activity level returned at 95%. From hospital days 6 to 26, he underwent apheresis with subsequent eculizumab (600 mg) every other day for 11 total doses. On hospital day 8, his platelet count began to spontaneously uptrend (Figure 2C). He progressed from anuria to oliguria but continued to require HD and has shown no evidence of further renal recovery after 5 months.
Methods
Patient population
Three consecutive patients with carfilzomib-induced aHUS at the University of Rochester Medical Center in 2017 and 2018 underwent genetic and functional complement pathway testing. A diagnosis of carfilzomib-induced aHUS was made using elevated LDH, decreased haptoglobin or elevated total bilirubin, evidence of schistocytes on PBS, anemia, thrombocytopenia, acute kidney injury, no evidence of infectious diarrhea, and active carfilzomib treatment.
Complement gene and functional testing
Testing was completed at the Molecular Otolaryngology and Renal Research Laboratories at the University of Iowa. TMA Functional Panel and Genetic Renal Panel v5 were performed on samples from each patient.
Results and discussion
Genetic and functional complement pathway testing was performed after resolution of acute illness. Patients 1 and 2 were found to harbor heterozygous CFHR3-CFHR1 deletions. No complement system gene mutations were detected in patient 3. CFH autoantibodies were negative and functional alternative complement pathway testing was unremarkable in all 3 patients.
Predisposing mutations to aHUS generally modulate alternative complement pathway function.6 A common homozygous deletion encompassing CFH-related protein 3 (CFHR3) and CFHR1 genes, found in 5.7% of persons of European-American and Finnish descent,7 is associated with aHUS.8,9 The absence of CFHR1 and CFHR3 enables uncontrolled complement activation on cell and tissue surfaces.10 However, heterozygous deletion is considered a common benign variant. In the setting of prolonged carfilzomib use, heterozygous CFHR3-CFHR1 deletion may represent a conditional mutation for the development of aHUS.
The effect of carfilzomib on complement function is poorly understood, but it may be related to proteasome-mediated gene regulation. Proteasome inhibition in vitro decreases CFH expression, thereby increasing the cytotoxic effect of complement activation.11,12 In this regard, carfilzomib may decrease CFH expression, diminishing alternative complement pathway inhibition, leaving the alternative complement pathway susceptible to dysfunction in the setting of innate impairment of CFH-related protein function.
Carfilzomib-induced aHUS typically manifests in patients who have undergone ≥1 cycle of treatment.3,4 However, development of disease is not related to total cumulative carfilzomib dose or underlying disease progression. Three potential etiologies, or combinations thereof, explain this delayed time-course: (1) stochastic carfilzomib triggering, (2) increasing CFH downregulation with long-term carfilzomib use, or (3) an additional triggering event.
In conclusion, development of carfilzomib-induced aHUS may be associated with heterozygous CFHR3-CFHR1 deletion. Further investigation of this relationship is warranted to determine the strength of any association and delineate potential mechanisms.
We advise early consideration of eculizumab, prior to the return of ADAMTS13 activity level and without concurrent apheresis, in patients with suspected carfilzomib-induced aHUS. Although the standard treatment duration of eculizumab in carfilzomib-induced aHUS has not been established, chronic eculizumab treatment is not necessary for all patients. Withdrawal may be considered after resolution of acute thrombocytopenia.
Authorship
Contribution: A.J.P. wrote the manuscript; B.L. conceived the project and provided oversight; and the final manuscript was reviewed and approved by both authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andrew Jay Portuguese, 601 Elmwood Ave, Rochester, NY 14642; e-mail: andrew_portuguese@urmc.rochester.edu.

