

Key Points
PN-1 constitutes an important endogenous mechanism of protection against lung fibrosis by limiting lung inflammation.
PN-1 is a cellular serpin that regulates PAR4-induced thrombin effects on inflammatory cells in the lung.

Abstract
Coagulation and fibrinolytic system deregulation has been implicated in the development of idiopathic pulmonary fibrosis, a devastating form of interstitial lung disease. We used intratracheal instillation of bleomycin to induce pulmonary fibrosis in mice and analyzed the role of serine protease inhibitor E2 (serpinE2)/protease nexin-1 (PN-1), a tissue serpin that exhibits anticoagulant and antifibrinolytic properties. PN-1 deficiency was associated, after bleomycin challenge, with a significant increase in mortality, as well as a marked increase in active thrombin in bronchoalveolar lavage fluids, an overexpression of extracellular matrix proteins, and an accumulation of inflammatory cells in the lungs. Bone marrow transplantation experiments showed that protective PN-1 was derived from hematopoietic cell compartment. A pharmacological strategy using the direct thrombin inhibitor argatroban reversed the deleterious effects of PN-1 deficiency. Concomitant deficiency of the thrombin receptor protease-activated receptor 4 (PAR4) abolished the deleterious effects of PN-1 deficiency in hematopoietic cells. These data demonstrate that prevention of thrombin signaling by PN-1 constitutes an important endogenous mechanism of protection against lung fibrosis and associated mortality. Our findings suggest that appropriate doses of thrombin inhibitors or PAR4 antagonists may provide benefit against progressive lung fibrosis with evidence of deregulated thrombin activity.
Introduction
Inflammation, coagulation, and fibrinolysis pathways are essential regulatory elements in normal tissue repair. Indeed, alterations in their equilibrium can result in unwanted tissue remodeling, resulting in fibrosis, a pathological situation often arising from nonresolving inflammation. Within the lung tissue, the links between these systems are tightly interrelated. Idiopathic pulmonary fibrosis (IPF) is a disease characterized by a strong inflammatory response in the early phase,1 an accumulation of fibroblasts and myofibroblasts, and the deposition of fibrin and extracellular matrix (ECM), such as collagen and fibronectin. This ECM deposition leads to changes in lung architecture and a loss of lung function.2 The fibrinolytic pathway has been shown to be involved in the development of pulmonary fibrosis. Indeed, IPF is characterized by increased antifibrinolytic activities in the lungs, leading to excessive deposition of intraalveolar fibrin.3,4 The coagulation cascade also plays a central role in influencing inflammatory and tissue repair mechanisms occurring during pulmonary fibrosis. Thrombin, a key serine protease of the coagulation cascade, not only contributes to fibrin formation and platelet activation, but also exerts direct cellular effects through activation of G protein–coupled protease-activated receptors (PARs), which may contribute to inflammation and fibrosis after tissue injury. Direct inhibition of thrombin has been reported to attenuate the fibrotic response in the bleomycin-induced lung fibrosis model.5,6 Moreover, PAR1 deficiency7 or pharmacological inhibition of PAR18 affords protection against bleomycin-induced lung fibrosis. Lastly, thrombin activation of PAR1 has been shown to promote fibroblast proliferation and differentiation into myofibroblasts.9
Serine protease inhibitors (serpins) are protease inhibitors that play central regulatory roles throughout the mammalian body. They serve as pivotal players in normal physiological functions and regulate protease activation in various systems, including coagulation, fibrinolysis, or inflammation. The role of serpins as central regulators of normal physiological functions is well illustrated by the number of diseases caused by serpin dysfunction.10 In inflammatory settings, unstable or unbalanced serpin levels can even be lethal, as in severe sepsis with disseminated intravascular coagulation.11 Plasminogen activator inhibitor-1 (PAI-1) is the principal serpin regulating fibrinolysis, whereas antithrombin (AT) and protease nexin-1 (PN-1; serpinE2) are the 2 principal known physiological inhibitors of thrombin. AT is a plasma serpin and therefore inhibits thrombin generated after vessel injury, whereas PN-1 is undetectable in the blood and acts essentially at the tissue/cellular level. Regarding lung injury, elevated expression of PAI-1 has been shown to reduce fibrinolysis in IPF,4,12 but PN-1 (serpinE2) may also contribute. Indeed, PN-1 inhibits thrombin,13,14 the plasminogen activators (PAs) urokinase-type PA and tissue PA, and plasmin.14-16 PN-1 has been shown to be expressed in the normal adult mouse lung17 and in normal and pathological human lungs.18 Its expression is upregulated in lung tissue, bronchoalveolar lavage fluids (BALFs), and lung fibroblasts from IPF patients, in whom it is able to block thrombin activity.19 Collectively, this may suggest a potential role for PN-1 in the pathophysiology of IPF. However, because PN-1 exerts both anticoagulant and antifibrinolytic activities, whether it plays a protective role in the cellular responses to lung injury remains undetermined. Moreover, because PN-1 is expressed not only by lung cells but also by inflammatory cells, the cellular origin of PN-1 involved in its potential role in the pathophysiology of pulmonary fibrosis also remains undetermined. To answer these questions, we have applied the well-established bleomycin mouse model of pulmonary fibrosis20 to PN-1+/+ and PN-1−/− mice.
Methods
Animals
PN-1–deficient (PN-1−/−)21 mice were from Denis Monard (Biomedical Research, Basel, Switzerland). PAR4-deficient (Par4−/−) mice were from Shaun Coughlin (Cardiovascular Research Institute, University of California San Francisco, San Francisco, CA).22 Experimental animals were 8 to 10 weeks of age. Heterozygous mating generated PN-1−/− and PN-1+/+ mice. Mice were bred and maintained in our own laboratory (Paris, France). All animals were genotyped by polymerase chain reaction (PCR). All animal experiments were performed in accordance with the Declaration of European Conventions for the Use and Care of Animals.
Bleomycin exposure
For each experiment, age- and weight-matched male mice were used. Mice were anesthetized by intraperitoneal injection of 100 mg/kg of ketamine and 10 mg/kg of xylazine in saline solution. On day 0, mice were intratracheally inoculated with bleomycin (2 mg/kg body weight) in 80 µL of saline (bleomycin bellon; Sanofi, Paris, France). Animals were euthanized on different days after instillation according to the experiment, and lungs were removed for further analysis. Saline-treated mice were used as controls. In additional series of experiments, argatroban (Arganova; LFB Biomedicaments, Les Ulis, France), a direct thrombin inhibitor, dissolved in the physiological serum in a volume of 50 µL was administered by intraperitoneal injection at a dose of 9 mg/kg body weight daily after bleomycin treatment.
BM transplantation
Male 5-week-old mice (PN-1−/− or their PN-1+/+ littermates or C57Bl/6 wild-type [WT] mice purchased from Jackson Laboratory, according to experiments) were subjected to 9 Gy of lethal total-body irradiation. The day after, irradiated mice were reconstituted by retroorbital injection of 4.5 × 106 cells of freshly isolated bone marrow (BM) from femurs and tibias of age- and sex-matched PN-1−/− or Par4−/− or PN-1−/−/Par4−/− or PN-1+/+ mice. Five weeks after injection of donor cells, mice were exposed to bleomycin treatment. Blood cells of recipient mice were genotyped to verify successful repopulation of donor cells.
BALF analysis and cell count
BALFs were collected in 1 mL of phosphate-buffered saline (PBS) supplemented with 1 mM of EDTA (Thermo-Fischer Scientific, Courtaboeuf, France). After cytocentrifugation, the cell pellet was reconstituted in 80 µL of PBS for counting at the hemocytometer, and the supernatant was stored at −20°C until analysis. The following enzyme-linked immunosorbent assays were used according to the manufacturer’s instructions: TGFβ1 (R&D Systems, Lille, France) and D-dimers (Interchim, Montluçon, France). Active thrombin was measured in BALFs by the fluorometric method. BALF aliquots (50 µL) were mixed at 37°C with 50 µL of buffer containing 20 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.5), 150 mM of sodium chloride, 0.1% human serum albumin, and 2 mM of thrombin substrate Pefafluor (H-D-CHA-Ala-Arg-AMC.2AcOH; Cryopep, Montpellier, France). Fluorescence was read at 390 to 460 nm on a spectrofluorometer and thrombin activity determined by extrapolation from a thrombin standard curve.
Measurement of collagen content in lung tissue
At 3, 6, and 9 days after bleomycin instillation, lung collagen was quantified in snap-frozen left lungs using the hydroxyproline assay kit (Sigma-Aldrich/Merck, Saint-Quentin Fallavier, France) according to manufacturer’s instruction and as described previously.8 Results are expressed in micrograms of hydroxyproline per milligram of lung.
Lung histology
After bleomycin or saline instillation, mice were euthanized, and lung tissue was harvested, fixed in 10% formaldehyde, embedded in paraffin, and cut into 10-µm thick sections. The sections were stained with Picro-Sirius red or Masson’s trichrome.
Reverse transcription and quantitative real-time PCR
Total RNA from snap-frozen left lung was extracted with Trizol and reverse transcribed using the Maxima reverse transcriptase (Thermo-Fischer Scientific, Courtaboeuf, France) as described by the manufacturer. The resulting complementary DNA was used as a template for quantitative PCR analysis of fibronectin, collagen1α2, collagen3α1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) messenger RNA (mRNA) expression in a LightCycler system with SYBR Green detection (Roche Applied Science, Meylan, France). The following run protocol for collagen1α2, collagen3α1, and fibronectin was used: denaturation: 95°C, 5 minutes; amplification and quantitation (40 cycles): 60°C, 10 minutes; 72°C, 30 minutes. Fluorescence of the samples was monitored continuously while the temperature was increased from 62°C to 95°C at a linear transition rate of 0.1°C s−1 and arrested by a final cooling step at 45°C. Collagen1α2 primers were: forward 5′-CATGTTCAGCTTTGTGGAC CT-3′ and reverse 5′-GCAGCTGACTTCAGGGATGT-3′; collagen3α1 primers were: forward 5′-CAATATGCCCACAGCCTTCTA-3′ and reverse 5′-CAGGAATGCCAGGAG GAC-3′; and fibronectin primers were: forward 5′-TGCACGATGATATGGAGAGC-3′ and reverse 5′-TGGGTGTCACCTGACTGAAC-3′. LightCycler run protocol for GAPDH was as follows: denaturation: 95°C, 5 minutes; amplification and quantitation (40 cycles): 65°C, 10 minutes; 72°C, 20 minutes. GAPDH primers were: forward 5′-TGTCCGTCGTGAATCTGAC-3′ and reverse 5′-CCTGCTTCACCACCTTCTTG-3′. Transcripts levels were normalized to GAPDH mRNA.
Immunoblot analysis
The lungs from PN-1+/+ and PN-1−/− mice were snap frozen and homogenized in lysis buffer, containing a cocktail of protease inhibitors. After centrifugation for 5 minutes at 15 000 × g at 4°C, total protein was measured using the BCA protein assay (Fisher Scientific, Illkirch, France), and 20 μg of proteins were loaded onto 10% acrylamide gels with sodium dodecyl sulfate polyacrylamide gel electrophoresis, electrophoresed, and transferred to nitrocellulose membranes (Hybond-ECL, Amersham Biosciences) that were saturated with blocking buffer containing 5% nonfat dry milk in 50 mM of phosphate, 150 mM of sodium chloride, and 0.1% Tween-20 (PBST) for 1 hour before incubation with mouse anti-human fibronectin (2.5 µg/mL; Santa Cruz, Heidelberg, Germany) or goat anti-mouse collagen1 (5 µg/mL; Southern Biotech, Birmingham, AL) and rabbit anti-GAPDH (2 μg/mL; Biolegend, London, United Kingdom) used for normalization in PBST overnight at 4°C. All blots were then washed with PBST and incubated for 2 hours at room temperature with horseradish peroxidase–conjugated secondary antibody. Immunoreactive bands were visualized by standard chemiluminescence (enhanced chemiluminescence reagent; GE Healthcare Life Sciences, Velizy-Villacoublay, France) and densitometric analysis performed by using Image J software.
Statistical analysis
Differences between experimental and control groups were analyzed using Student t test or Mann-Whitney U test. Differences between groups were analyzed by 1-way analysis of variance followed by Tukey’s multiple comparisons test or Kruskal Wallis test, as appropriate. The log-rank test/Kaplan-Meier test was used for statistical analysis of survival rates. P < .05 was considered statistically significant.
Results
PN-1−/− mice show a higher susceptibility to bleomycin-induced mortality
A single bleomycin instillation in mice is considered a relevant model for patients with active IPF.23 Using this model, we observed that PN-1−/− mice displayed a significant decrease in survival compared with their PN-1+/+ littermates (Figure 1). Nine days after bleomycin instillation, 60% of PN-1−/− mice had died, relative to 25% of PN-1+/+ controls; 14 days after bleomycin treatment, 90% of PN-1−/− mice had died, relative to ∼50% of PN-1+/+ controls. PN-1 thus protects against mortality in the bleomycin model.
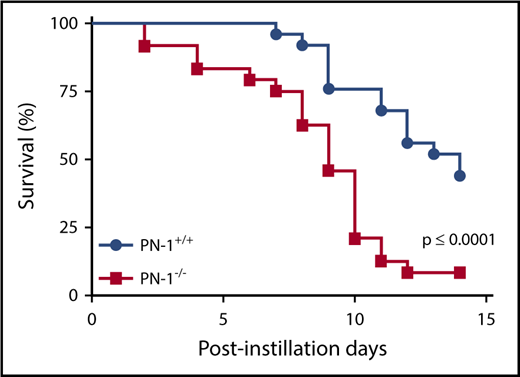
Increased mortality in bleomycin-injured PN-1−/−mice.PN-1−/− mice and their PN-1+/+ littermates were subjected to bleomycin-induced lung injury (2 mg/kg) by intratracheal instillation. Percentages of surviving mice were plotted over a 14-day period after bleomycin treatment. Log-rank test was used to compare the difference between PN-1−/− and PN-1+/+ mice (n = 25 per group; P ≤ .0001). The proportion of survival was determined based on euthanasia criteria. Animals that lost 20% of their body weight were euthanized.
Increased mortality in bleomycin-injured PN-1−/−mice.PN-1−/− mice and their PN-1+/+ littermates were subjected to bleomycin-induced lung injury (2 mg/kg) by intratracheal instillation. Percentages of surviving mice were plotted over a 14-day period after bleomycin treatment. Log-rank test was used to compare the difference between PN-1−/− and PN-1+/+ mice (n = 25 per group; P ≤ .0001). The proportion of survival was determined based on euthanasia criteria. Animals that lost 20% of their body weight were euthanized.
PN-1−/− mice have increased inflammatory and coagulation responses in the lung after bleomycin treatment
To understand the biological effect of PN-1 during IPF, we examined the inflammatory and coagulation responses to intratracheal instillation of bleomycin in both PN-1−/− and PN-1+/+ mice. It is known that an acute inflammatory response in the early phase after bleomycin treatment can contribute to fibrotic progression.1 Therefore, we studied the inflammatory response of mice early after bleomycin treatment by analyzing cell counts in BALFs from PN-1+/+ and PN-1−/− mice. White blood cells (WBCs) were detectable in the BALFs of PN-1+/+ and PN-1−/− mice treated with physiological serum (sham); however, no significant difference was observed in the total number of WBCs between these 2 groups, ruling out an increased basal inflammatory state in the lungs of PN-1−/− vs PN-1+/+ mice (Figure 2A). Although bleomycin treatment increased the WBCs in BALFs of all mice, the increase was ∼2 times greater in PN-1−/− mice compared with PN-1+/+ mice (Figure 2A). Differential cell count of WBCs was performed by flow cytometry analysis and showed that lymphocytes, monocytes, and neutrophils were significantly more prevalent after bleomycin treatment in the lungs of PN-1−/− mice compared with PN-1+/+ mice (supplemental Figure 1). Similarly, platelets were significantly more prevalent in the lungs of PN-1−/− mice than in PN-1+/+ mice after bleomycin treatment (Figure 2B). However, the red blood cell counts in the BALFs were similar between PN-1−/− and PN-1+/+ bleomycin-instilled mice (Figure 2C). These findings suggest that PN-1 deficiency accelerates bleomycin-induced lung fibrosis by affecting the inflammatory reaction in response to lung injury.
![Figure 2. Accentuated pulmonary inflammatory and coagulation responses in lungs of bleomycin-injured PN-1−/− mice. PN-1−/− and their PN-1+/+ littermates were subjected to bleomycin-induced lung injury (BLM; 2 mg/kg) or physiological serum (sham) by intratracheal instillation for 4 to 5 days. BALFs were collected from lung tissues and the number of WBCs (A), platelets (B), and red blood cells (RBCs) (C) were determined by counting in the hemocytometer. Thrombin activity was measured by the fluorometric method (D) and D-dimer by enzyme-linked immunosorbent assay (E). Data (mean ± standard error of the mean [SEM]; n = 6-8 per group) were analyzed by 1-way analysis of variance with a Tukey’s multiple comparison test. *P < .05, **P < .01, ***P < .001, ****P < .0001 vs respective control. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/18/10.1182_bloodadvances.2018018283/4/m_advances018283f2.png?Expires=1769148770&Signature=nlfqTb9JXJ9cuVAiUI0-9l97tYiTO7Y7dnnTgl1IbvwbcRgZpKCMD7h0DtRNJdcBAPQABEYh-IgxxDa7e193BsufPlUZRqVJCnyRuDuWbA-Kt4UnVYMXMg~71WC0uizvxmCGNGq8iFTmj1KT68mgBfSlYMoIR-8Ez6Kl7DS77r8WEyBDQzUcjiOsx~mxuOUBXTdfTFnqZEsuC2rVeD7EBMWUNRNhPdsws55Aisfd4~KskDldhArZXjOjvReUopVLaW656apjsm8rJOpqaPFklHNJbAmYGG7Qy--8yUBorKAEYyF3lwT2K~ssKPbvhMHs9P1apWSr8XHQOaH1-KeWog__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Accentuated pulmonary inflammatory and coagulation responses in lungs of bleomycin-injured PN-1−/−mice.PN-1−/− and their PN-1+/+ littermates were subjected to bleomycin-induced lung injury (BLM; 2 mg/kg) or physiological serum (sham) by intratracheal instillation for 4 to 5 days. BALFs were collected from lung tissues and the number of WBCs (A), platelets (B), and red blood cells (RBCs) (C) were determined by counting in the hemocytometer. Thrombin activity was measured by the fluorometric method (D) and D-dimer by enzyme-linked immunosorbent assay (E). Data (mean ± standard error of the mean [SEM]; n = 6-8 per group) were analyzed by 1-way analysis of variance with a Tukey’s multiple comparison test. *P < .05, **P < .01, ***P < .001, ****P < .0001 vs respective control. ns, not significant.
Accentuated pulmonary inflammatory and coagulation responses in lungs of bleomycin-injured PN-1−/−mice.PN-1−/− and their PN-1+/+ littermates were subjected to bleomycin-induced lung injury (BLM; 2 mg/kg) or physiological serum (sham) by intratracheal instillation for 4 to 5 days. BALFs were collected from lung tissues and the number of WBCs (A), platelets (B), and red blood cells (RBCs) (C) were determined by counting in the hemocytometer. Thrombin activity was measured by the fluorometric method (D) and D-dimer by enzyme-linked immunosorbent assay (E). Data (mean ± standard error of the mean [SEM]; n = 6-8 per group) were analyzed by 1-way analysis of variance with a Tukey’s multiple comparison test. *P < .05, **P < .01, ***P < .001, ****P < .0001 vs respective control. ns, not significant.
Coagulation activation accompanies inflammation in the early stages of lung injury.5 Consistent with previous reports, we observed that thrombin levels increased in BALFs soon after bleomycin instillation (Figure 2D). Thrombin activity was threefold higher in BALFs from bleomycin-treated PN-1−/− mice relative to PN-1+/+ mice 4 days after instillation, with a corresponding increase in D-dimer levels, indicating an ongoing process of fibrin formation and degradation (Figure 2E). PN-1 thus makes an important contribution to thrombin inhibition during bleomycin-induced lung injury.
PN-1−/− mice have increased lung fibrosis after bleomycin treatment
We also analyzed the fibrotic response to bleomycin injury. Because a majority of PN-1−/− mice die soon after bleomycin treatment, we chose day 3 and day 9 for fibrosis assessment. Collagen content was significantly increased in PN-1−/− mice relative to littermate controls at both 6 and 9 days after bleomycin instillation, as assessed by Masson’s trichrome and Sirius red staining of lung sections (Figure 3A) and by measuring hydroxyproline levels in snap-frozen lungs (Figure 3B). Collagen content was also significantly increased in PN-1+/+ mice 14 days after bleomycin treatment, indicating that fibrosis development was delayed compared with PN-1−/− mice, as assessed by the time course of hydroxyproline in the lungs of PN-1+/+ mice (supplemental Figure 2).
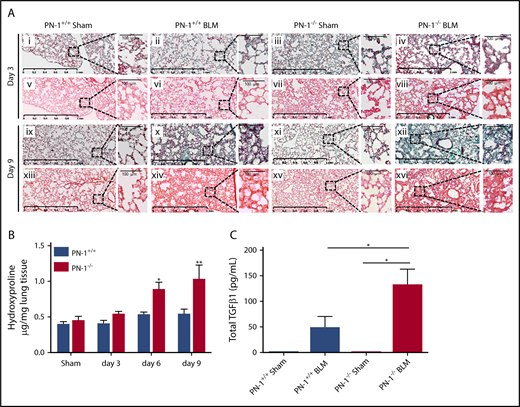
Accentuated pulmonary fibrosis and increased TGFβ1 in PN-1−/−mice. Lungs from PN-1−/− mice and their PN-1+/+ littermates were harvested at the indicated time points after bleomycin-induced lung injury (BLM) instillation for the following analyses. (A) Masson’s trichrome (i-iv, ix-xii) and Sirius red (v-viii, xiii-xvi) staining of lung sections from PN-1+/+ and PN-1−/− mice 3 and 9 days after saline (sham) or BLM treatment. Representative images are shown. Scale bars, 1 mm (main images) and 100 μm (enlargements). (B) Hydroxyproline contents in lung tissues from PN-1−/− and PN-1+/+ mice were measured 3, 6, and 9 days after bleomycin treatment vs saline treatment (sham). Data (mean ± SEM; n = 4-5 per group) were analyzed by 2-tailed Mann-Whitney U test. (C) Total TGFβ levels were measured by enzyme-linked immunosorbent assay in BALFs collected 9 days after saline (sham) or BLM treatment. Data (mean ± SEM; n = 10-13 per group) were analyzed by Kruskall-Wallis test with Dunn’s multiple comparison test. *P < .05, **P < .01 vs sham.
Accentuated pulmonary fibrosis and increased TGFβ1 in PN-1−/−mice. Lungs from PN-1−/− mice and their PN-1+/+ littermates were harvested at the indicated time points after bleomycin-induced lung injury (BLM) instillation for the following analyses. (A) Masson’s trichrome (i-iv, ix-xii) and Sirius red (v-viii, xiii-xvi) staining of lung sections from PN-1+/+ and PN-1−/− mice 3 and 9 days after saline (sham) or BLM treatment. Representative images are shown. Scale bars, 1 mm (main images) and 100 μm (enlargements). (B) Hydroxyproline contents in lung tissues from PN-1−/− and PN-1+/+ mice were measured 3, 6, and 9 days after bleomycin treatment vs saline treatment (sham). Data (mean ± SEM; n = 4-5 per group) were analyzed by 2-tailed Mann-Whitney U test. (C) Total TGFβ levels were measured by enzyme-linked immunosorbent assay in BALFs collected 9 days after saline (sham) or BLM treatment. Data (mean ± SEM; n = 10-13 per group) were analyzed by Kruskall-Wallis test with Dunn’s multiple comparison test. *P < .05, **P < .01 vs sham.
Because TGFβ1 is a critical player in ECM deposition during pulmonary fibrosis,24 we assessed TGFβ1 levels in BALFs of PN-1−/− and control mice. Total TGFβ1 was undetectable in control BALFs from both PN-1+/+ and PN-1−/− mice but increased significantly relative to sham animals 9 days after bleomycin instillation only in PN-1−/− mice (Figure 3C). Accordingly, active TGFβ1 was detectable only in BALFs from bleomycin-treated PN-1−/− mice (12.7 ± 4.4 pg/mL).
Accentuated fibrosis in PN-1−/− mice was further supported by increased mRNA levels for ECM components in lung extracts compared with PN-1+/+ mice (Figure 4A-C). Expression of collagen3α1 and collagen1α2 was increased by six- and 12-fold, respectively, compared with sham PN-1−/− mice 9 days after bleomycin instillation (Figure 4A-B). This was accompanied by a 26-fold increase in fibronectin expression relative to sham PN-1+/+ mice (Figure 4C). These results were confirmed at the protein level by immunoblotting of ECM proteins (Figure 4D-F). Thus, PN-1 suppresses the expression and deposition of ECM proteins after a bleomycin challenge.

Accentuated overexpression of matrix extracellular proteins in lungs of bleomycin-injured PN-1−/−mice. Lungs from PN-1−/− mice and their PN-1+/+ littermates were harvested at the indicated time points after bleomycin-induced lung injury (BLM) instillation for the following analyses. Quantitative reverse transcription PCR analysis of collagen3α1 (col3α1) (A), col1α2 (B), and fibronectin (C) mRNA expressions in lung tissues. GAPDH was used as an internal control (n = 4 per group). (D) Western blot analysis of type 1 collagen (col 1) and fibronectin. GAPDH was used as a loading control. Densitometric analysis of the corresponding western blot analyses is expressed for fibronectin (E) and col 1 (F) (n = 6-10 per group). For panels A-C and E-F, data represent mean ± SEM and were analyzed by 2-tailed Mann-Whitney U test. *P < .05, **P < .01 vs respective sham.
Accentuated overexpression of matrix extracellular proteins in lungs of bleomycin-injured PN-1−/−mice. Lungs from PN-1−/− mice and their PN-1+/+ littermates were harvested at the indicated time points after bleomycin-induced lung injury (BLM) instillation for the following analyses. Quantitative reverse transcription PCR analysis of collagen3α1 (col3α1) (A), col1α2 (B), and fibronectin (C) mRNA expressions in lung tissues. GAPDH was used as an internal control (n = 4 per group). (D) Western blot analysis of type 1 collagen (col 1) and fibronectin. GAPDH was used as a loading control. Densitometric analysis of the corresponding western blot analyses is expressed for fibronectin (E) and col 1 (F) (n = 6-10 per group). For panels A-C and E-F, data represent mean ± SEM and were analyzed by 2-tailed Mann-Whitney U test. *P < .05, **P < .01 vs respective sham.
Protective role of PN-1 expression by hematopoietic cells in bleomycin-induced lung injury
PN-1 is expressed in hematopoietic cells, including platelets, monocytes, and neutrophils, as well as in resident cells in lung. We addressed the source of protective PN-1 by performing BM transplantation experiments. Irradiated PN-1−/− and PN-1+/+ mice underwent transplantation with either PN-1−/− or PN-1+/+ BM to create 4 chimeric groups of mice, which were allowed to recover for 5 weeks and then challenged with bleomycin. Transplantation of PN-1+/+ BM into PN-1−/− mice rescued PN-1−/− recipients from bleomycin-induced lethality, whereas transplantation of PN-1−/− BM into PN-1+/+ mice increased the mortality of PN-1+/+ recipients, highlighting the protective role of hematopoietic cell–derived PN-1 in bleomycin-induced injury (Figure 5A). Hematopoietic PN-1 also limited the development of fibrosis, because collagen depositions in the lungs of PN-1−/− mice transplanted with PN-1+/+ BM cells and in the lungs of PN-1+/+ mice transplanted with PN-1−/− BM cells were decreased and increased, respectively, relative to homotypic transplant recipients (Figure 5B). These results argue that PN-1 provided by hematopoietic cells is thus an important protective factor in bleomycin-induced lung injury.
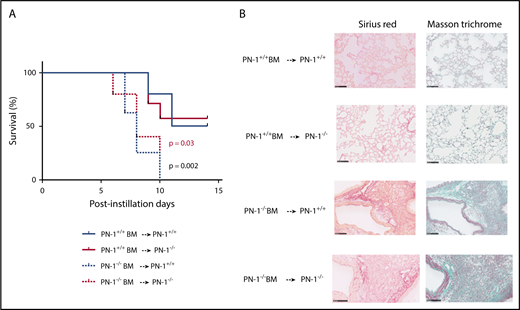
Protective effect of PN-1 from BM cells in bleomycin-injured chimeric mice.PN-1−/− mice and their PN-1+/+ littermates were irradiated and underwent transplantation with BM from appropriated mice and allowed to recover for 5 weeks before bleomycin-induced lung injury. PN-1+/+ BM → PN-1+/+: PN-1+/+ mice receiving PN-1+/+ BM transplants (n = 10). PN-1−/− BM → PN-1+/+: PN-1+/+ mice receiving PN-1−/− BM transplants (n = 8). PN-1+/+ BM → PN-1−/−: PN-1−/− mice receiving PN-1+/+ BM transplants (n = 7). PN-1−/− BM → PN-1−/−: PN-1−/− mice receiving PN-1−/− BM transplants (n = 5). (A) Percentages of surviving mice undergoing transplantation were plotted over a 14-day period after bleomycin treatment. Log-rank test was used to compare the difference between similar recipient mice. P = .03 for PN-1+/+ BM → PN-1−/− vs PN-1−/− BM → PN-1−/−, and P = .002 for PN-1+/+ BM → PN-1+/+ vs PN-1−/− BM → PN-1+/+. (B) Masson’s trichrome and Sirius red stainings of lung withdrawn the day of euthanasia from PN-1+/+ and PN-1−/− chimeric mice. Representative images are shown. Scale bars, 100 µm.
Protective effect of PN-1 from BM cells in bleomycin-injured chimeric mice.PN-1−/− mice and their PN-1+/+ littermates were irradiated and underwent transplantation with BM from appropriated mice and allowed to recover for 5 weeks before bleomycin-induced lung injury. PN-1+/+ BM → PN-1+/+: PN-1+/+ mice receiving PN-1+/+ BM transplants (n = 10). PN-1−/− BM → PN-1+/+: PN-1+/+ mice receiving PN-1−/− BM transplants (n = 8). PN-1+/+ BM → PN-1−/−: PN-1−/− mice receiving PN-1+/+ BM transplants (n = 7). PN-1−/− BM → PN-1−/−: PN-1−/− mice receiving PN-1−/− BM transplants (n = 5). (A) Percentages of surviving mice undergoing transplantation were plotted over a 14-day period after bleomycin treatment. Log-rank test was used to compare the difference between similar recipient mice. P = .03 for PN-1+/+ BM → PN-1−/− vs PN-1−/− BM → PN-1−/−, and P = .002 for PN-1+/+ BM → PN-1+/+ vs PN-1−/− BM → PN-1+/+. (B) Masson’s trichrome and Sirius red stainings of lung withdrawn the day of euthanasia from PN-1+/+ and PN-1−/− chimeric mice. Representative images are shown. Scale bars, 100 µm.
PN-1 protects against bleomycin-induced lung injury by inhibiting thrombin
Thrombin is known to exert potent effects on inflammatory cells. Exacerbated coagulation and inflammatory responses in lungs of bleomycin-treated PN-1−/− mice may thus be explained by uncontrolled thrombin activity. We therefore assessed the effect of thrombin inhibition on the deleterious effect of PN-1 deficiency in the bleomycin model. PN-1−/− mice were injected daily after bleomycin instillation with the direct inhibitor of thrombin, argatroban, and 2 ex vivo assays were used to monitor thrombin-dependent coagulation: activated partial thromboplastin time and thrombin time. Confirming effective thrombin inhibition, the mean thrombin time of argatroban-treated PN-1−/− mice was prolonged from 20.4 ± 1.3 to ≥300 seconds (P < .03) and the mean activated partial thromboplastin time from 44.2 ± 6.4 to 152.8 ± 17.8 seconds (P < .03), compared with vehicle argatroban treatment, which completely inhibited thrombin in BALFs of PN-1−/− mice (Figure 6A). Interestingly, argatroban significantly improved the survival of PN-1−/− mice (Figure 6B). Argatroban treatment did not influence the numbers of WBCs (0.41 ± 0.03 × 109/L vs 0.31 ± 0.07 × 109/L) or platelets (176 ± 34 × 109/L vs 144 ± 46 × 109/L) in BALFs of PN-1−/− mice administered intratracheal saline (sham). In contrast, it significantly decreased the numbers of both platelets (Figure 6C) and WBCs (Figure 6D) in BALFs of bleomycin-injured PN-1−/− mice. The protective function of PN-1 during bleomycin-induced lung injury can therefore be attributed to its ability to inhibit thrombin. We also assessed the effect of thrombin inhibition on the survival of bleomycin-treated PN-1+/+ mice. In contrast to the results obtained with PN-1−/− mice, argatroban treatment significantly accelerated the mortality of bleomycin-treated PN-1+/+ mice (supplemental Figure 3A). Such accelerated mortality was explained by bleeding in lungs, as assessed by quantification of hemoglobin revealing an abnormal presence of blood in BALFs of PN-1+/+ mice compared with their control (supplemental Figure 3B).

Protective effect of thrombin inhibition by argatroban in bleomycin-injured PN-1−/−mice.PN-1−/− mice were subjected to bleomycin-induced lung injury (BLM; 2 mg/kg) and treated daily with argatroban (9 mg/kg intraperitoneally) . (A) Thrombin activity was measured by a fluorometric method. Data (mean ± SEM; n = 9-11 per group) were analyzed by Kruskall-Wallis test with Dunn’s multiple comparison test. (B) Percentages of surviving PN-1−/− mice were plotted over a 14-day period. Log-rank test was used to compare the difference between PN-1−/− mice with BLM and PN-1−/− mice with BLM plus argatroban (PN-1−/− BLM: n = 10; PN-1−/− BLM + argatroban: n = 15; P = .02). The number of platelets (C) and WBCs (D) in BALFs were counted in the hemocytometer. Data (mean ± SEM; n = 6-9 per group) were analyzed by 1-way analysis of variance with Tukey’s multiple comparison test. *P < .05, ***P < .001, ****P < .0001.
Protective effect of thrombin inhibition by argatroban in bleomycin-injured PN-1−/−mice.PN-1−/− mice were subjected to bleomycin-induced lung injury (BLM; 2 mg/kg) and treated daily with argatroban (9 mg/kg intraperitoneally) . (A) Thrombin activity was measured by a fluorometric method. Data (mean ± SEM; n = 9-11 per group) were analyzed by Kruskall-Wallis test with Dunn’s multiple comparison test. (B) Percentages of surviving PN-1−/− mice were plotted over a 14-day period. Log-rank test was used to compare the difference between PN-1−/− mice with BLM and PN-1−/− mice with BLM plus argatroban (PN-1−/− BLM: n = 10; PN-1−/− BLM + argatroban: n = 15; P = .02). The number of platelets (C) and WBCs (D) in BALFs were counted in the hemocytometer. Data (mean ± SEM; n = 6-9 per group) were analyzed by 1-way analysis of variance with Tukey’s multiple comparison test. *P < .05, ***P < .001, ****P < .0001.
PAR4 deficiency abolishes the deleterious effect of PN-1 deficiency on hematopoietic cells in bleomycin-induced pulmonary fibrosis
The hematopoietic origin of protective PN-1 in this model suggested that thrombin might exert its pathogenic action within the hematopoietic compartment. Because thrombin mediates platelet activation via PAR4 in mice, we investigated whether hematopoietic PAR4 was a target for the pathogenic effects of deregulated thrombin activity in PN-1−/− mice. Irradiated C57Bl/6 WT mice underwent transplantation with BM from PN-1−/−/Par4+/+, PN-1+/+/Par4−/−, PN-1−/−/Par4−/−, or PN-1+/+/Par4+/+ mice to create 4 groups of chimeric mice. WT mice receiving PN-1+/+/Par4−/− BM transplants did not differ significantly in survival rates from WT mice receiving PN-1+/+/Par4+/+ BM transplants (Figure 7). As previously observed, WT mice receiving PN-1−/−/Par4+/+ BM transplants showed a dramatic decrease in survival compared with PN-1+/+ mice receiving PN-1+/+/Par4+/+ BM transplants. Interestingly, WT mice receiving PN-1−/−/Par4−/− BM transplants displayed survival rates similar to WT mice receiving PN-1+/+/Par4+/+ BM transplants (Figure 7). Thus, deregulated thrombin activity in PN-1–deficient mice increases mortality from bleomycin-induced pulmonary fibrosis by inducing PAR4 signaling in hematopoietic cells.
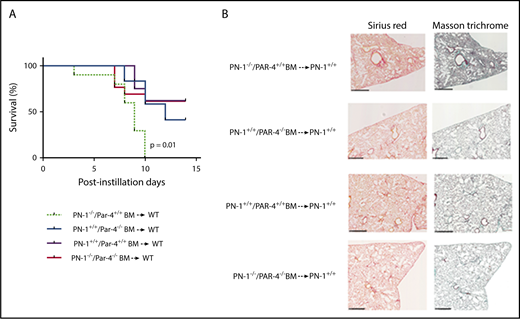
Protective effect of PAR4 expressed on hematopoietic cells in bleomycin-injured lungs. WT mice were irradiated and underwent transplantation with PN-1−/−/Par4+/+ or PN-1+/+/Par4−/− or PN-1+/+/Par4+/+ or PN-1−/−/Par4−/− BM and allowed to recover for 5 weeks before bleomycin-induced lung injury. PN-1−/−/Par4+/+ BM → WT (n = 10). PN-1+/+/Par4−/− BM → WT (n = 12). PN-1+/+/Par4+/+ BM → WT (n = 8). PN-1−/−/Par4−/− BM → WT (n = 12). (A) Percentages of surviving mice undergoing transplantation were plotted over a 14-day period after bleomycin treatment. Log-rank test was used to compare the difference between recipient mice: P = .01 for PN-1−/−/Par4+/+ BM → WT vs PN-1+/+/Par4+/+ BM → WT. (B) Masson’s trichrome and Sirius red stainings of lung withdrawn the day of euthanasia from WT chimeric mice. Representative images are shown. Scale bars, 500 µm.
Protective effect of PAR4 expressed on hematopoietic cells in bleomycin-injured lungs. WT mice were irradiated and underwent transplantation with PN-1−/−/Par4+/+ or PN-1+/+/Par4−/− or PN-1+/+/Par4+/+ or PN-1−/−/Par4−/− BM and allowed to recover for 5 weeks before bleomycin-induced lung injury. PN-1−/−/Par4+/+ BM → WT (n = 10). PN-1+/+/Par4−/− BM → WT (n = 12). PN-1+/+/Par4+/+ BM → WT (n = 8). PN-1−/−/Par4−/− BM → WT (n = 12). (A) Percentages of surviving mice undergoing transplantation were plotted over a 14-day period after bleomycin treatment. Log-rank test was used to compare the difference between recipient mice: P = .01 for PN-1−/−/Par4+/+ BM → WT vs PN-1+/+/Par4+/+ BM → WT. (B) Masson’s trichrome and Sirius red stainings of lung withdrawn the day of euthanasia from WT chimeric mice. Representative images are shown. Scale bars, 500 µm.
Discussion
IPF is a form of interstitial pneumonia of unknown pathogenic mechanisms. However, in the early stages after lung injury, closely linked pathways such as inflammatory reactions, coagulation, and fibrinolysis are known to be involved in disease progression.1 Evidence of inflammation is consistently found in lungs of patients with IPF, because inflammatory cells were significantly more numerous in the lungs of the rapid decliner group of patients with IPF.25 Moreover, a bleomycin challenge has been shown to induce an acute inflammatory reaction in the lung with a pronounced influx of leukocytes in the alveolar interstitium and alveolar spaces,23 and mice treated with bleomycin exhibited platelet sequestration within the lungs, supporting a role for platelets in lung injury.26 Our present data provide additional arguments for the relative importance of inflammation in the development of IPF. Indeed, we demonstrate here that deficiency in PN-1, a serpin characterized by its anticoagulant and antifibrinolytic properties, resulted in an increased inflammatory response during lung injury, as assessed by increased inflammatory cell counts and increased TGFβ levels in the BALFs of lung-injured PN-1−/− mice. Interestingly, inflammatory cells were recently shown to accumulate in lungs from aged PN-1−/− mice.27 PN-1 deficiency is thus closely related to the uncontrolled or overexuberant inflammatory reactions responsible for lung injury in the early stages of the disease.
Besides inflammation, impaired fibrinolysis has also been implicated in the development of IPF, indicating that fibrinolytic proteases regulate fibrotic processes in injured lungs. Several studies have indeed demonstrated a protective role of PAI-1 deficiency in the bleomycin model of lung fibrosis.12,28,29 This protective effect results from an enhancement of the fibrinolytic activity. Interestingly, although PN-1 can also regulate fibrinolysis by inhibiting plasmin generation and activity, our data showed that, in contrast to PAI-1 deficiency, PN-1 deficiency aggravates bleomycin-induced pulmonary fibrosis. This increased fibrotic phenotype displayed by PN-1−/− mice was revealed by higher collagen and fibronectin content in the injured lung tissue, indicating that the antifibrinolytic property of PN-1 was not important for IPF development. Indeed, in these PN-1−/− mice challenged with bleomycin, fibrinolytic activity could still be regulated by PAI-1, a serpin known to be overexpressed during bleomycin-induced lung injury.30 Moreover, biochemical studies comparing the rate constants for inhibition of tissue PA or urokinase-type PA by PN-1 and PAI-1 indicate that PN-1 is a less efficient inhibitor of fibrinolysis than PAI-1.31
The overexpression of extracellular matrix proteins we observed in PN-1−/− mice seems to contradict our previously reported results showing that overexpression of PN-1 increases fibronectin expression in fibroblasts from healthy donors.19 It should be noted that these previous data did not show any significant change for collagen expression. These apparently conflicting results can be explained by the fact that our present data were obtained in an in vivo mouse model, where both overexpressed PAI-1 and PN-1 act together in the lung tissue, which is not the case in vitro using transfected cultured cells.
The fact that PN-1−/− mice experience worse outcomes after bleomycin injury with increased mortality and fibrosis underlines the importance of the anticoagulant effect of PN-1 during lung injury. Increased procoagulant activity has been observed in the lungs of IPF patients.4 It has also been shown that alveolar macrophages and, to a lesser extent, alveolar type 2 cells are the main sources of locally produced tissue factor and therefore the sources of thrombin in bleomycin-injured lungs,32 indicating that coagulation proteases regulate fibrotic processes in injured lungs. Here, we clearly demonstrate that the protective role of PN-1 results from its ability to inhibit thrombin, as evidenced by the rescue of PN-1−/− mice after treatment with the direct thrombin inhibitor argatroban. Our data indicate that thrombin level was too high in BALFs of lung-injured PN-1−/− mice and therefore responsible for detrimental outcomes of the bleomycin treatment. It thus seems that after argatroban treatment, thrombin level was reduced to a level sufficient to maintain hemostasis in the lungs of PN-1−/− mice to avoid bleeding and at the same time low enough to improve outcomes after bleomycin treatment. In contrast, argatroban clearly accelerated mortality of bleomycin-treated PN-1+/+ mice because of lung bleeding. Such detrimental outcome of argatroban treatment in bleomycin-treated PN-1+/+ mice was also explained by thrombin level. Indeed, thrombin level measured in BALFs was 3 times lower in argatroban-treated PN-1+/+ mice than in argatroban-treated PN-1−/− mice. Therefore, thrombin level was reduced to a level too low to maintain hemostasis in the lungs of argatroban-treated PN-1+/+ mice, resulting in lung bleeding.
The importance of thrombin regulation during lung injury was also shown in a previous study demonstrating, in the bleomycin pulmonary fibrosis model, the anti-inflammatory and antifibrotic effects of dabigatran, another small molecule inhibiting thrombin.33 Because serpins are the major regulators of antiproteases in the lung, our data point out the importance of serpins able to finely regulate thrombin activity in this organ. Besides PN-1, AT is an efficient inhibitor of thrombin. However, AT has been essentially shown to be involved in sepsis-related lung dysfunction or in ischemia reperfusion of grafted lungs, but no data are available regarding pulmonary fibrosis.
We showed previously that PN-1 expression is upregulated in lung tissue, BALFs, and lung fibroblasts from IPF patients.19 At first sight, these previous results may seem to contradict our present data showing that PN-1−/− mice have aggravated bleomycin-induced pulmonary fibrosis. However, if we consider that increased levels of thrombin activity are not accompanied by a similar increase in the levels of endogenous PN-1, we may propose that PN-1 overexpression represents an early protective response to lung injury that becomes overwhelmed when thrombin generation is too great.
In addition to its procoagulant effect, thrombin exerts various cellular effects that contribute to inflammatory and fibrotic processes in the lung.5,34,35 The cellular effects displayed by thrombin are mainly mediated by PAR1 and PAR4. PAR1 is involved in tissue repair and fibrosis through stimulation of endothelial cells and regulation of fibroblast proliferation and differentiation36 and therefore influences principally stromal cells of the lungs. PAR4 plays a role in thrombin-induced recruitment of polymorphonuclear cells37 and in activation of platelets in mice.38 Previous data did not show any protection against bleomycin-induced pulmonary fibrosis in PAR4−/− mice.39 Indeed, the authors did not see any difference in the severity of fibrotic changes or in profibrotic gene expression like fibronectin or collagen between WT and PAR4−/− mice. This is in agreement with our data showing no difference between WT mice receiving PN-1+/+/Par4+/+ vs PN-1+/+/Par4−/− BM transplants. However, the concomitant deficiency of PN-1 and PAR4 in BM cells can reverse the deleterious effect of isolated PN-1 deficiency, suggesting that unchecked thrombin promotes inflammatory PAR4 signaling in this context. Our data thus show that PN-1 expressed by BM cells is essential to prevent thrombin-induced aggravation of lung injury. We clearly show that protective PN-1 comes from BM cells, but the specific cell type expressing it remains to be identified. Because platelets are a major source of PN-1,40 we can speculate they play an important role in the protective function of PN-1. To address this possibility, we will need to generate PN-1–deficient mice specifically in the megakaryocyte or myeloid lineage and characterize lung fibrosis. This is beyond the scope of our work, but our data clearly indicate that PN-1 plays a key role in lung injury. An increasing number of data provide evidence of a protective role played by PN-1 in the different tissues where it is expressed.41 This is also the case in lungs, where we show that thrombin can significantly contribute to the progression and impact of lung injury if not regulated by PN-1. Interestingly, PAR4 inhibition is now considered an effective and safe potential new antithrombotic drug candidate.42 Given the need for novel therapeutic targets in IPF, and considering our present data, PAR4 antagonists that are currently under development for clinical use in the treatment of cardiovascular diseases should also be considered in the treatment of patients with active lung fibrosis.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank J. Miloradovic for excellent technical assistance and J. Marchal-Somme for her advice concerning the bleomycin-induced lung injury model in mice.
D.F. and Y.B. were the recipients of a PhD fellowship and “Aide au retour en France” program from the “Fondation pour la Recherche Médicale,” respectively. This work was supported by INSERM, Université Paris Diderot, and Agence Nationale de la Recherche (ANR-13-BSV3-0011).
Authorship
Contribution: M.-C.B., D.F., and Y.B. conceived and designed the study; D.F., L.V., L.I., R.M., and K.A. performed most of the experiments; V.A., Y.B., S.G., and L.C. performed the bone marrow transplantation experiments; V.A., M.J.-P., and E.C. provided intellectual discussion and editorial advice; M.-C.B., Y.B., and D.F. analyzed the data and wrote the paper; and all authors approved the submission of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yacine Boulaftali, Unité INSERM U1148- LVTS, CHU Xavier Bichat, 46 rue Henri Huchard, 75877 Paris Cedex 18, France; e-mail: yacine.boulaftali@inserm.fr.

