Key Points
High MTV immediately before lymphodepletion and CD30.CAR-T cell infusion is associated with shorter PFS.
High pretherapy MTV is associated with high circulating CD3+PD-1+ T cells after CD30.CAR-T infusion.

Abstract
Our group has recently demonstrated that chimeric antigen receptor T-cell therapy targeting the CD30 antigen (CD30.CAR-T) is highly effective in patients with relapsed and refractory (r/r) classical Hodgkin lymphoma (cHL). Despite high rates of clinical response, relapses and progression were observed in a subset of patients. The objective of this study was to characterize clinical and correlative factors associated with progression-free survival (PFS) after CD30.CAR-T cell therapy. We evaluated correlatives in 27 patients with r/r cHL treated with lymphodepletion and CD30.CAR-T cells. With a median follow-up of 9.5 months, 17 patients (63%) progressed, with a median PFS of 352 days (95% confidence interval: 116-not reached), and 2 patients died (7%) with a median overall survival of not reached. High metabolic tumor volume (MTV, >60 mL) immediately before lymphodepletion and CD30.CAR-T cell infusion was associated with inferior PFS (log rank, P = .02), which persisted after adjusting for lymphodepletion and CAR-T dose (log rank, P = .01 and P = .006, respectively). In contrast, receiving bridging therapy, response to bridging therapy, CD30.CAR-T expansion/persistence, and percentage of CD3+PD-1+ lymphocytes over the first 6 weeks of therapy were not associated with differences in PFS. In summary, this study reports an association between high baseline MTV immediately before lymphodepletion and CD30.CAR-T cell infusion and worse PFS in patients with r/r cHL. This trial was registered at www.clinicaltrials.gov as #NCT02690545.
Introduction
Chimeric antigen receptor T (CAR-T) cells represent an effective therapy option for the treatment of patients with relapsed/refractory (r/r) B-cell lymphoma and acute lymphoblastic leukemia, and most recently for patients with r/r multiple myeloma.1-4 Our group recently evaluated the efficacy of CD30.CAR-T therapy in a cohort of heavily pretreated patients with r/r classical Hodgkin lymphoma (cHL).5 In this multicenter phase 1/2 study, patients were treated with escalating doses of CD30.CAR-T cells after lymphodepletion with bendamustine, bendamustine and fludarabine, or cyclophosphamide and fludarabine. In patients with measurable disease, the objective response rate (ORR) was 62%, the complete response (CR) rate was 51%, and the 1-year progression-free survival (PFS) was 36%. It remains elusive whether improved clinical response or duration of response to CD30.CAR-T therapy can be predicted in patients with r/r cHL. Thus, studies to evaluate variables that impact the response to CD30.CAR-T cell therapy are currently needed.
CAR-T cells targeting the CD19 antigen (CD19.CAR-T) have been studied more extensively in diffuse large B-cell lymphoma (DLBCL) and other aggressive B-cell lymphomas. Several recent studies have examined possible clinical and correlative factors associated with ORR and PFS after CD19.CAR-T therapy. In a real-world analysis of axicabtagene ciloleucel (axi-cel), lower baseline C-reactive protein and higher absolute lymphocyte count at time of apheresis were associated with improved ORR.6 Outcomes from tisagenlecleucel (tis-cel) in the JULIET trial have shown that elevated lactate dehydrogenase levels before CAR-T infusion and c-MYC expression by immunohistochemistry are associated with inferior ORR and PFS.7,8 Additionally, PFS after CD19.CAR-T has been shown to increase with higher intensity lymphodepleting chemotherapy, which is thought to promote a more favorable cytokine profile prior to CAR-T administration.9
Volumetric tumor burden has also been an area of active investigation in evaluating responses to CD19.CAR-T therapy. In the pivotal ZUMA-1 trial, the presence of bulky disease (>10 cm) was associated with a nonstatistically significant inferior ORR.1 However, given the limitations to assess tumor volume by measuring the sum of the product of diameter (SPD) approach in lymphoma, additional approaches including functional imaging modalities have been evaluated. Baseline metabolic tumor volume (MTV) from 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)/computed tomography (CT) before CD19.CAR-T cell therapy for DLBCL has been shown in multiple recent studies to predict PFS and overall survival (OS).10,11 Controversy remains regarding the appropriate threshold for determining low vs high MTV, and currently there is no standardized value accepted. It is likely that threshold values will need to be tailored to lymphoma subtypes and/or phase of therapy.10-14 For cHL, threshold values for MTV have varied from 8 to 225 mL depending on the clinical setting of the analysis (initial diagnosis, before autologous stem cell transplant, and relapsed disease).15-20
In this study, we examined data from a single center cohort of patients with r/r cHL who received CD30.CAR-T cell therapy to identify variables associated with improved PFS.
Patients and methods
Patient population
Patients in this analysis were enrolled in a phase 1/2 clinical trial (ClinicalTrials.gov identifier: NCT02690545) of lymphodepletion followed by autologous CD30.CAR-T cell therapy at the University of North Carolina (UNC) between August 2016 and June 2020. All patients provided written informed consent. Eligibility included any patient with a CD30-positive lymphoma at the time of lymphodepletion and CD30.CAR-T administration. CD30.CAR-T cells were manufactured at the UNC Good Manufacturing Practice–compliant facility (IND14688). Tolerability and initial clinical outcomes in the cHL cohort have previously been reported.5 Bridging chemotherapy was allowed but not required. Patients achieving a CR with bridging therapy were allowed to receive CD30.CAR-T cells and were included in this analysis. Lymphodepletion consisted of bendamustine 90 mg/m2 per day for 3 days in the initial 8 patients treated and then changed to bendamustine 70 mg/m2 per day and fludarabine 30 mg/m2 per day for 3 days in the remaining 19 patients. A dose escalation strategy was used with a CAR-T dose of 1 × 108 CAR-Ts/m2 in the first 3 adult and pediatric patients, followed by a dose of 2 × 108 CAR-Ts/m2 in the remaining patients. This article reports on all patients with r/r cHL treated with CD30.CAR-T at UNC through June 2020. This includes updated outcome and correlative data in the 25 patients previously published from the UNC cohort and 2 additional patients not previously published.
Clinical response and relapse assessments
Disease response was determined by PET/CT imaging at 6 weeks after CAR-T cell therapy using the Lugano Criteria.21 PFS was defined as days from CD30.CAR-T infusion to relapse, progression, or death. Patients without events were censored at their last follow-up date or at the cutoff date of 1 November 2020, whichever was earlier.
CAR-T cell expansion and programmed cell death protein 1 expression
Expansion and persistence of CD30.CAR-T cells in vivo was determined by quantitative polymerase chain reaction (qPCR) from peripheral blood samples.5,22 Programmed cell death protein 1 (PD-1) expression was determined by flow cytometry (MIH1 clone) on CD45+CD3+ gated mononuclear cells (supplemental Figure 1). Peripheral blood time points for both qPCR and flow cytometry included the following: prelymphodepletion and 1, 2, 3, 4, and 6 weeks after CD30.CAR-T cell infusion.5
MTV measurement
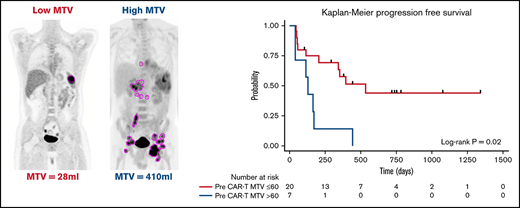
All patients had baseline 18F-FDG PET/CT scans before lymphodepletion and CD30.CAR-T cell infusion. In patients who had a 18F-FDG PET/CT prior to bridging therapy, these scans were evaluated for MTV to provide a tumor burden comparison before and after bridging therapy. 18F-FDG PET/CT scans were evaluated using MIM 7.0.5 (MIM Software, Cleveland, OH). MTV was computed using the threshold tool in MIM with a threshold 41% of the maximum standardized uptake value (SUV), or SUVmax, as described elsewhere.10-14 The lesion was manually selected and the region of interest increased until all voxels at least 41% of the SUVmax (Figure 1). MTV for each patient was determined by summing the volumes of lesions with voxels at least 41% of the SUVmax.
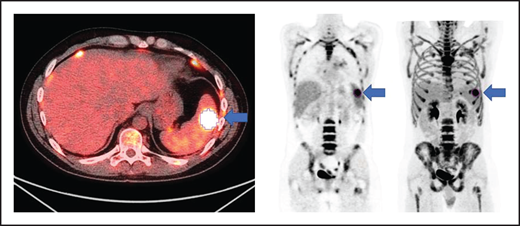
Axial fused, coronal PET-only, and maximum intensity projection images. Representative lesion segmented using 41% of SUVmax as cutoff (blue arrows).
Axial fused, coronal PET-only, and maximum intensity projection images. Representative lesion segmented using 41% of SUVmax as cutoff (blue arrows).
Statistical analyses
Biomarkers of interest were analyzed using a complete-case, twofold, cross-validating technique in which the cohort was randomly divided into twofolds of equal size. Biomarker cutoffs were determined by choosing a cutoff in each fold to maximize the Z-score for comparing PFS, using the log-rank test, between the high biomarker (above cutoff) subgroup and the low biomarker (below or equal to cutoff) subgroup in that fold. An average of the cutoffs from each fold was then applied to the full cohort to compare PFS between biomarker high and low subgroups, reported as a 2-sided log-rank P value. For each fold, average cutoffs were obtained using unadjusted analyses and with either adjustment for lymphodepletion or CD30.CAR-T cell dose. PFS was summarized using the Kaplan-Meier method. Median survival time and 1-year survival probability were calculated.
Area under curve (AUC) calculations for both CD30.CAR-T cell expansion and PD-1 expression on CD3+ lymphocytes were performed with log transformation of data across time points using the trapezoidal rule. Missing values were imputed from values of the closest 2 other time points, assuming a log-linear trend. All AUC cutoffs were evaluated based on the twofold cross-validation procedure with the log-transformed data. Correlations between MTV and either CD30.CAR-T cell expansion or PD-1 expression on CD3+ lymphocytes were evaluated by calculating a Pearson correlation r statistic and corresponding P value.
Results
Patient characteristics
A total of 27 patients were treated with CD30.CAR-T cell therapy for r/r cHL at UNC between August 2016 and June 2020 (Table 1). At the time of enrollment, the median age of patients was 33 years (range, 15-67 years). Most patients were male (n = 18, 67%), had nodular sclerosis type histology (n = 18, 67%), and had stage III/IV disease at the time of diagnosis (n = 18, 67%). Patients were heavily pretreated with a median of 8 prior lines of therapy (range, 3-23 lines of therapy). Eighty-one percent of the treated patients had progressed despite a prior autologous stem cell transplant (n = 22) and 37% had progressed after an allogeneic stem cell transplant (n = 10).
Characteristics of bridging therapy, lymphodepletion, and CD30.CAR-T cell therapy
Seventy percent (19 patients) of the cohort received bridging therapy between blood collection for CAR-T manufacturing and lymphodepletion (Table 1). Bridging therapy agents included the following: bendamustine (n = 8), bendamustine plus everolimus (n = 1), bendamustine plus nivolumab (n = 1), anti–PD-1 monoclonal antibody therapy (nivolumab or pembrolizumab, n = 4), other salvage chemotherapy (n = 3), everolimus (n = 1), or brentuximab vedotin (n = 1). Local radiation was administered in combination with systemic therapy in 2 patients. The ORR to bridging therapy was 58% with 5 patients (26%) achieving a CR. Patients who achieved a CR with bridging therapy were still eligible for lymphodepletion and CD30.CAR-T cell therapy.
Lymphodepletion was administered to all patients treated with CD30.CAR-T cells. Bendamustine was the lymphodepletion regimen for the first 8 patients treated (30%), and a combination of bendamustine and fludarabine was used for the remaining 19 patients of the cohort (70%). Five patients (19%) were treated with CD30.CAR-T cells at a dose of 1 × 108 cells/m2 as part of dose escalation. The remaining 22 patients (81%) were treated at a dose of 2 × 108 cells/m2. Following lymphodepletion and CD30.CAR-T cells, the ORR at 6 weeks was 71% with 18 patients (67%) achieving a CR. Two patients had stable disease (SD) and subsequently progressed, and 6 patients (22%) had progressive disease (PD). Overall, 17 patients (63%) in this cohort developed progression with a median PFS of 352 days (95% confidence interval [CI]: 116-not reached), and 2 patients died (7%) with a median OS of not reached (Figure 2).
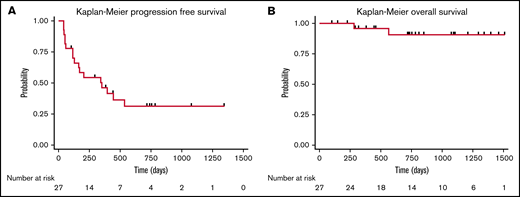
Cohort survival. PFS (A) and OS (B) for all patients treated with CD30.CAR-T cell therapy.
Cohort survival. PFS (A) and OS (B) for all patients treated with CD30.CAR-T cell therapy.
Impact of MTV on PFS
The median time from 18F-FDG PET/CT scan to CD30.CAR-T cell infusion was 12 days (range, 4-35 days). MTV calculated from 18F-FDG PET/CT scans obtained after bridging therapy (if applicable) and before lymphodepletion and CD30.CAR-T cell infusion were evaluated by a twofold, cross-validating analysis. This revealed a trend toward inferior PFS in patients with high pre–CAR-T MTV (P = .07; Table 2). After adjusting for lymphodepletion, high pre–CAR-T MTV was associated with inferior PFS (P = .03). Analyzing pre–CAR-T MTV with a cutoff of 60 mL revealed that high MTV > 60 mL was associated with inferior PFS in both the unadjusted model and after adjustments for lymphodepletion and CAR-T dose (P = .02, 0.01, and 0.006, respectively; Figure 3A). The 1-year PFS estimate was 14% (95% CI: 1%-46%) and 58% (95% CI: 33%-76%) in the high and low baseline MTV groups, respectively.

PFS by baseline MTV. (A) PFS by high (blue) vs low (red) MTV, using a cutoff of 60 mL. (B) PFS by change to MTV with bridging therapy using a cutoff of 60 mL. Low MTV to low MTV (red), high MTV to low MTV (blue), and high MTV to high MTV (light green). *Pairwise P value low to low MTV compared with high to high MTV.
PFS by baseline MTV. (A) PFS by high (blue) vs low (red) MTV, using a cutoff of 60 mL. (B) PFS by change to MTV with bridging therapy using a cutoff of 60 mL. Low MTV to low MTV (red), high MTV to low MTV (blue), and high MTV to high MTV (light green). *Pairwise P value low to low MTV compared with high to high MTV.
The impact of bridging therapy on MTV and subsequent PFS after CD30.CAR-T cell therapy was also evaluated. Patients who had low MTV (≤60 mL) before bridging therapy and continued to have low MTV before CD30.CAR-T cell infusion had the best PFS (1-year PFS 75%; 95% CI: 13%-96%). Patients who had high MTV (>60 mL) that improved with bridging therapy to low MTV before CD30.CAR-T cell infusion had intermediate PFS (1-year PFS 40%; 95% CI: 5%-75%). Finally, patients with high MTV that did not improve to low MTV with bridging therapy had poor PFS (1-year PFS 0%; P = .047; Figure 3B).
Impact of bridging therapy, CD30.CAR T expansion, and PD-1 expression on PFS
There was no difference in PFS in the unadjusted estimate between the 70% of patients who received bridging therapy and the 30% of those who did not receive bridging therapy (P = .35; Table 2). Evaluating patients who received bridging therapy (n = 19), there was no difference in PFS in patients who achieved a clinical response (CR or partial response) compared with no clinical response (SD or PD; P = .76). In both analyses, there was no significant impact of adjustments for either lymphodepletion or CD30.CAR-T dose.
CD30.CAR-T cell expansion and persistence was evaluated by quantifying CAR expression using qPCR from mononuclear cells isolated from the peripheral blood. Samples were collected every week for 6 weeks after cell infusion (supplemental Figure 2). The peak of CAR T-cell expansion was determined as the highest value up to 6 weeks after infusion. Peak CD30.CAR-T expansion was not associated with a difference in PFS in this cohort (P = .35; Table 2). Additionally, CD30.CAR-T persistence was estimated by measuring the AUC obtained by the qPCR values obtained over a period of 6 weeks after infusion. The AUC of CD30.CAR-T expansion from cell infusion (day 0) to day 42 was not associated with a difference in PFS (P = .23).
The percentage of PD-1–expressing CD3+ lymphocytes in the peripheral blood was evaluated before CD30.CAR-T infusion and then weekly after the infusion for up to 6 weeks (supplemental Figure 3). PD-1 expression at time points before CD30.CAR-T infusion or after the CD30.CAR-T infusion was not associated with difference in PFS (Table 2). The CD3+PD-1+ AUC from days 0 to 42, representing the burden of PD-1 expression over this time, was not statistically associated with differences in PFS (P = .16). The AUC of PD-1 expression between days 14 and 42 after cell infusion, representing the impact of delayed PD-1 expression after CD30.CAR-T infusion, was not associated with PFS (P = .43).
Correlation of CD30.CAR-T expansion and PD-1 expression with MTV
We evaluated whether pre–CAR-T MTV correlated with CD30.CAR-T expansion and persistence and PD-1 expression on CD3+ lymphocytes over the first 6 weeks of therapy. MTV positively correlated with the AUC for PD-1 expression from days 0 to 42 (r = 0.441, P = .045) and the AUC for PD-1 expression from days 14 to 42 (r = 0.551, P = .01). In contrast, MTV was not correlated with peak CD30.CAR-T expansion (r = −0.05, P = .81), AUC for CD30.CAR-T cells from days 0 to 42 (r = 0.005, P = .98), or AUC for CD30.CAR-T cells from days 14 to 42 (r = −0.05, P = .68; Table 3). Additionally, MTV was not correlated with pre–CAR-T cytokine levels (interleukin 15 [IL-15], IL-6, tumor necrosis factor α [TNFα], thymus and activation regulated chemokine) or peak cytokine levels after CD30.CAR-T infusion.
Discussion
Although patients with r/r HL achieved a high objective response rate after lymphodepletion and CD30.CAR-T cell therapy, disease progression within 1-year of therapy occurred in more than 60% of patients.5 In this analysis, we evaluated clinical and correlative data to identify patients with prolonged responses after CD30.CAR-T cell therapy. Increased MTV before lymphodepletion and CD30.CAR-T cell infusion, reflecting the amount of tumor volume with high glucose metabolism, was associated with inferior PFS. In contrast, having received bridging therapy, response to bridging therapy, and CD30.CAR-T expansion and persistence were not associated with differences in PFS.
The use of MTV for prognostication in response to treatment has been studied in a variety of lymphoma subtypes.10-20 A recent meta-analysis of 27 studies evaluating the impact of MTV on survival outcomes in lymphoma reported a pooled hazard ratio of 3.05 (95% CI: 2.55-3.64; P < .001) for PFS.16 In cHL specifically, MTV has been shown in both limited and advanced stage disease to be predictive of PFS at the time of diagnosis.15,17,20 In r/r cHL, high MTV at time of relapse and before AutoSCT were predictive of worse PFS.18,19 In this study, we demonstrated a strong association between MTV before lymphodepletion and PFS after CD30.CAR-T cell therapy. Interestingly, neither objective response by Lugano criteria nor Deauville score before lymphodepletion and CD30.CAR-T cell therapy were associated with differences in PFS (supplemental Figure 4). Furthermore, high MTV > 60 mL, when applied to 18F-FDG PET/CT scans before bridging therapy, retained predictive value in determining shorter PFS with CD30.CAR-T cell therapy (P = .04; supplemental Figure 5).
Although high MTV may be indicative of poor long-term outcomes regardless of the intervention, we observed that improving MTV with bridging therapy in r/r cHL before lymphodepletion and CD30.CAR-T cell infusion had a positive impact on the PFS after therapy. Our observation underscores the clinical benefit of reducing tumor metabolic burden with bridging therapy in cHL before lymphodepletion and CAR-T cell infusion. Furthermore, given the known impact of high MTV on outcomes with CD19.CAR-T cell therapy in DLBCL, our study supports the value of minimizing metabolic tumor burden before CAR-T cell therapies in lymphoma.
High MTV in r/r cHL may be indicative not only of higher tumor burden but also of a more prominent immunosuppressive tumor microenvironment (TME) that further hampers the effector function of CD30.CAR-T cells. A recent analysis of nutrient partitioning in the TME showed that myeloid cells, including myeloid-derived suppressor cells, have the highest capacity for glucose uptake within the TME.23 Given the extensive presence of both Th2 T cells and immunosuppressive innate immune cells that characterize the TME of cHL, the immune infiltrate may account for a significant portion the FDG avidity in cHL. Therefore, changes in MTV in cHL potentially reflect remodeling of the TME. Specifically, a low MTV may be a surrogate for a more favorable TME, depleted of inhibitory regulatory T cells, Th2 cells, and immunosuppressive innate cells, which would facilitate local CD30.CAR-T penetration and effector function.
We observed that high MTV correlated with higher frequency of CD3+PD-1+ T cells in the peripheral blood measured as AUC from days 0 to 42 and more strongly as AUC from days 14 to 42 after CD30.CAR-T cell infusions. We were unable to establish if PD-1+ CD30.CAR-T cells had a similar distribution. PD-1 expression on CD3+ peripheral lymphocytes is a dynamic measurement that can represent both T-cell activation and T-cell exhaustion in different clinical and temporal contexts.24,25 A few studies have evaluated PD-1 expression on both CD3+ lymphocytes and CD19.CAR-T cells directly. PD-1 expression has been associated with exhausted CAR-T cells and subsequent progressive disease. Wang et al26 described a limited set of patients with DLBCL who progressed after CD19/20.CAR-T cell therapy, were found to have increased intratumoral CD3+PD-1+ T cells, and subsequently responded to anti–PD-1 therapies. Chong et al27,28 described the ability to rescue exhausted CD19.CAR-T cells with anti–PD-1 therapy leading to CD19.CAR-T re-expansion and clinical response. In patients treated with CD30.CAR-T cell therapy, the association of high MTV with higher circulating CD3+PD1+ T cells could suggest a more global immune impaired milieu is predominant in patients with shorter PFS. This observation paves the rationale of combining CD30.CAR-T cell therapy and anti–PD-1 therapy in a time sequential manner. For instance, anti–PD-1 therapy at the time of relapse after CD30.CAR-T cell therapy is currently being investigated at our institution (NCT04134325) with the rationale that CD30.CAR-T cell therapies may reshape the TME and favor the emergence of endogenous tumor-specific T cells that can be reinvigorated by checkpoint blockade.
In our current analysis of CD30.CAR-T cells in r/r cHL, peak expansion, AUC days 0 to 42, or AUC days 14 to 42 of CD30.CAR-T cells, did not associate with differences in PFS. The impact of CAR-T expansion and persistence on clinical outcomes has yielded varying results. In ZUMA-1, which analyzed axi-cel in DLBCL, peak expansion of CD19.CAR-T cells was associated with clinical response.1 However, in JULIET, which analyzed tisa-cel in DLBCL, both peak expansion of CD19.CAR-T cells and persistence measured as AUC of CAR-T expansion were not associated with response.2 Unfortunately, CAR-T expansion and persistence in the peripheral blood represents only a conduit for true CAR-T effector function occurring within tumors. Future studies investigating local CAR-T penetration, expansion, and persistence are warranted.
This study has some limitations. The exploratory nature of the study, despite being derived from a prospective clinical trial, resulted in a heterogenous cohort of individuals with varying lymphodepletion regimens and CD30.CAR-T cell doses. Although we accounted for differences in lymphodepletion and CD30.CAR-T cell dose with survival analysis adjustments, unmeasured confounding remains a concern with this study. The small patient numbers also limited our ability to build a multivariable predictive model for long-term responders. However, the single variable analyses in this study are poised to guide future investigations.
This study reports an important predictive biomarker of long-term response for CD30.CAR-T cell therapy in r/r cHL. The impact of MTV on outcomes should be evaluated in subsequent prospective trials. Importantly, the potential impact of bridging therapy modulating MTV and TME composition should be an area of investigation in future studies evaluating CD30.CAR-T cell therapy.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute grant RO1HL114564 (to B.S.), the University Cancer Research Fund at the Lineberger Comprehensive Cancer Center (to B.S. and G.D.), a Stand Up 2 Cancer grant (G.D.), a Lymphoma Research Foundation Career Development Grant (N.G.), National Institutes of Health grant T32 CA211056-05 (to T.J.V.), an American Society of Clinical Oncology Young Investigator Award (to T.J.V.), and an American Association for Cancer Research/AstraZeneca Lymphoma research fellowship (to T.J.V.).
Authorship
Contribution: T.J.V., N.G., C.J.C., G.D., and B.S. conducted the research and wrote the manuscript; J.O. performed the MTV calculations; T.J.V., B.Z., J.O., G.H., A.K., C.D., J. Smith, A.I., A.W.B., G.D., B.S., N.G., and J. Serody discussed and interpreted the results; and N.G., J.K.M. S.P., T.C.S., and J. Serody wrote the clinical protocol.
Conflict-of-interest disclosure: The University of North Carolina (UNC) has a research collaboration with Tessa Therapeutics. Competing interests of authors from UNC are managed in accordance with institutional policies. B.S., G.D., and J. Serody have a pending patent concerning the product presented in this manuscript. J. Serody additionally has an international patent filed regarding the use of STING agonists to enhance CAR-T cell therapy. B.S., G.D., and N.G. have consulted for Tessa Therapeutics. N.G. has received research grant support from Genentech. T.J.V. has received research grant support from AstraZeneca. K.M. has consulted for Vesselon. The remaining authors declare no competing financial interests.
Correspondence: Natalie Grover, Lineberger Comprehensive Cancer Center, University of North Carolina–Chapel Hill, 170 Manning Dr, CB# 7305, Chapel Hill, NC 27599; e-mail: natalie_grover@med.unc.edu.