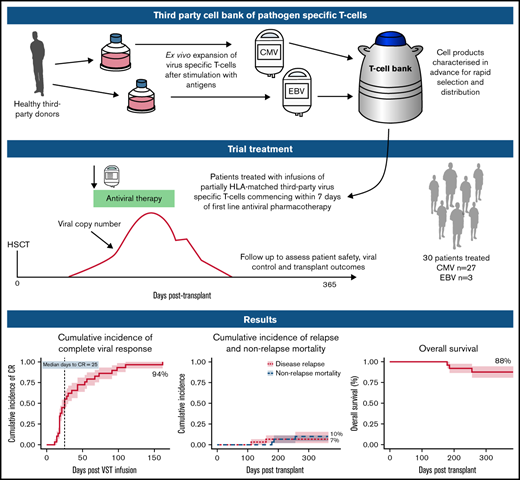
Key Points
Early use of third-party virus-specific T-cells is safe and leads to high rates of viral control and excellent outcomes in HSCT.
Virological clearance is associated with recovery of virus-specific immunity, in particular CD8+ effector memory T-cells.

Abstract
Virus-specific T-cells (VSTs) from third-party donors mediate short- and long-term antiviral effects in allogeneic hematopoietic stem cell transplant (HSCT) recipients with relapsed or refractory viral infections. We investigated early administration of third-party VSTs, together with antiviral therapy in patients requiring treatment for first cytomegalovirus (CMV) or Epstein-Barr virus (EBV) infection. Thirty HSCT patients were treated with 1 to 4 VST infusions (2 × 107 cells/m2; CMV n=27, EBV n=3) at a median of 4 days after initiation of antiviral treatment. The overall viral response rate was 100%, with a complete response (CR) rate of 94%. Of the 28 patients who achieved a CR, 23 remained virus PCR negative (n=9) or below quantitation limit (n=14) for the duration of follow-up. Four patients had brief episodes of quantifiable reactivation not requiring additional therapy, and one required a second infusion after initial CR, remaining PCR negative thereafter. All 3 patients treated for EBV post-transplant lymphoproliferative disorder achieved sustained CR. Rates of aGVHD and cGVHD after infusion were 13% and 23%, respectively. There were no serious infusion-related adverse events. VST infusion was associated with rapid recovery of CD8+CD45RA−CD62L− and a slower recovery of CD4+CD45RA−CD62L− effector memory T-cells; CMV-specific T-cells comprised up to 13% of CD8+ cells. At 1 year post-transplant, non-relapse mortality was 10%, cumulative incidence of relapse was 7%, overall survival was 88% and 25 of 27 patients had ECOG status of 0 or 1. Early administration of third-party VSTs in conjunction with antiviral treatment appears safe and leads to excellent viral control and clinical outcomes. Registered on Australian New Zealand Clinical Trials Registry as #ACTRN12618000343202.
Introduction
Opportunistic viral infections remain a significant cause of morbidity and mortality after allogeneic stem cell transplantation.1-3 HLA mismatch between donor and recipient, T-cell depletion, and immunosuppression aimed at minimizing graft-versus-host disease (GVHD) all impair the recovery of adaptive immunity and predispose to viral infection.4-6 Conventional antivirals have several drawbacks, including cost, toxicities, and development of resistance, and are not available for all infections. Coinfection with multiple viruses is frequent and adversely affects survival.1 Unless underlying immune deficiency can be corrected, patients are at high risk of recurrent viremia or tissue infection requiring prolonged or repeated antiviral treatment and resulting in organ toxicity and poor quality of life.
Adoptive T-cell therapy with virus-specific T-cells (VSTs) is an approach that has been used to enhance immune recovery.7-9 VSTs may be transplant donor–derived7-9 or generated from a third-party donor.10-12 Because adoptive T-cell therapy with third-party VSTs requires only partial HLA matching, a single VST product exerting an antiviral effect through a common HLA molecule can be used to treat any patient with the relevant infection who also expresses that HLA molecule.11,13 A small bank of donors selected for common HLA molecules can cover >95% of transplant recipients.13 Its cells can be cryopreserved and rapidly accessed when required. In patients with viral infection refractory to standard treatment, third-party VSTs have led to excellent viral control up to 12 months after infusion, with no significant safety issues.12
In this study, we assessed the safety and efficacy of third-party VSTs given together with standard-of-care antivirals at the time of initial viral infection requiring treatment after transplant. We hypothesized that by reconstituting immunity earlier than in previous studies, this treatment may avoid the negative consequences of prolonged antiviral administration in some transplant recipients and lead to rapid control of viral infection.
Methods
Study design and participants
A prospective, single-arm, multicenter, phase 1 clinical trial was performed in allogeneic stem cell transplant recipients with viral replication and/or tissue infection with cytomegalovirus (CMV) or Epstein-Barr virus (EBV). The trial was open to those with adenovirus infection; 1 patient was recruited for this indication who was later deemed ineligible. Eligibility for trial participation required that patients receive VSTs within 7 days of commencing standard antiviral therapy (supplemental Methods 1). No patient received letermovir prophylaxis. Exclusion criteria included active grade 2 to 4 acute GVHD (aGVHD), treatment with >1 mg/kg per day of prednisone or equivalent, treatment with antilymphocyte globulin, deranged hepatic or renal function, and Eastern Cooperative Oncology Group performance status (ECOG) >3. Detailed inclusion and exclusion criteria are included in supplemental Methods 1. Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki. The study was approved by the institutional research ethics committee at each site before the recruitment of patients.
Manufacture of T-cells from third-party donors
T-cell products were expanded from granulocyte colony-stimulating factor–primed apheresis product from healthy stem cell donors. Twelve donors with common HLA types and serological evidence of past exposure to CMV or EBV were recruited from Westmead Hospital according to standard assessment of eligibility for allogeneic donation. HLA typing was performed by the Australian Red Cross Blood Service and was resolved to 4 digits at class I and II loci in 10 of 12 donors and low resolution in 2 of 12. A bank of 17 monovalent VST products was generated (7 CMV specific, 5 EBV specific, and 5 adenovirus specific under good manufacturing practice conditions at the Sydney Cellular Therapies Laboratory, Westmead Hospital. CMV pp65 and EBV consensus peptides were added to donor blood or hemopoietic cell collection on day 1, followed by selection of CD137-expressing cells using antibody and magnetic bead selection (Miltenyi, Bergisch Gladbach, Germany) on day 2. The CD137− population was irradiated and peptide pulsed. The fractions were co-cultured for up to 11 days in the presence of cytokines (interleukin-2 [IL-2], IL-7 and IL-15), and then cryopreserved. Before release, all products underwent quality control testing as described in supplemental Methods 2.
Treatment and VST donor-recipient matching
Each dose consisted of 2.0 × 107/m2 partially HLA-matched CMV- or EBV-specific VSTs. VSTs were matched to the recipient at a minimum of 1 of 6 HLA antigens (HLA-A, -B, and -DRB1) shared between donor VSTs and recipient. VSTs were selected based on an algorithm that preferred the highest number of HLA matches with antiviral activity through the shared HLA antigen(s), and secondary preference to the product with the highest proportions of virus-specific responses through shared allele(s). After administration of the cells, up to 3 additional VST infusions could be given in the event of persistent viral replication detected ≥2 weeks after first VST infusion. In the event that a second infusion was required, the same product was used if it was available in the bank and had led to at least a partial virological response after initial administration. Concomitant standard-of-care antiviral treatment was administered to all patients according to local institutional policy, with choice of antiviral at the discretion of the treating physician.
Management of immunosuppression while on trial
Patients were excluded from the trial if they had received a corticosteroid dose of >1 mg/kg prednisolone or methylprednisolone (or equivalent in other preparations) within 72 hours before VST infusion. Patients receiving lower doses had their doses maintained without change for 72 hours before and at least 7 days after VST infusion. The rate of corticosteroid reduction after VST infusion did not exceed 10% per week of the dose given at the time of infusion. Doses of calcineurin inhibitors (cyclosporine, tacrolimus, or sirolimus) were not weaned in the absence of calcineurin toxicity until the dose of corticosteroids was <0.25 mg/kg.
Outcomes and follow-up
Patients were monitored at regular intervals for evidence of clinical and virological response, immune cell recovery, and toxicity. Regular clinical reviews were conducted and included a full history, physical examination, clinical GVHD assessment, and virus disease-specific assessment. The review cycle was reset after each infusion in the case of multiple infusions. Patients were actively followed up for 6 months to assess viral control and for correlative study samples. Patients were followed up for relapse and death until the time of data cutoff.
Adverse events were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03. The primary end point was safety of the VST infusion. Secondary end points are listed in supplemental Methods 3. GVHD was graded according to standard criteria for aGVHD14 and cGVHD.15
Viral infection
A complete virological response to treatment (CR) was defined as a decrease in viral load to levels undetectable by quantitative polymerase chain reaction (PCR), with resolution of clinical symptoms and signs. A partial response (PR) was defined as a decrease in viral load of at least 50% from baseline, with alleviation of clinical symptoms and signs. The entire area under the curve (AUC) of viral load was calculated to reflect the burden of viremia over time.16 All viral load measurements across different assays and analyzers were standardized to IUs per milliliter, using recognized conversion factors (supplemental Methods 4). Detectable CMV and EBV at any level of quantitation was considered positive (including positive results below the level of quantitation). CMV tissue infection was defined according to Chemaly et al.17 The response of patients with EBV-associated post-transplant lymphoproliferative disease was monitored according to both viral titers and International Lymphoma Working Group 2017 Consensus Criteria.18
Immune monitoring
Post-infusion immune monitoring was performed on batched peripheral blood mononuclear cells at specific time points for each patient by using spectral flow cytometry directed at 20 different surface markers, allowing for the delineation of 32 cellular populations of interest (supplemental Methods 5). Data were acquired with a FACSymphony Flow Cytometer (BD Biosciences, San Jose, CA). HLA-restricted antigen specificity in postinfusion CD8+ T-cell populations was measured using phycoerythrin-conjugated, virus-specific iTAg MHC class I human tetramers (MBL International, Woburn, MA) where available. CMV-specific T-cell immune recovery was monitored by interferon-γ (IFN-γ) enzyme-linked immunospot (Elispot) assay, as previously described.19
Cellular persistence
Persistence of transferred cells in the peripheral blood of recipients was assessed with a highly sensitive microchimerism assay on a droplet digital platform, as previously described.20 DNA extracted from peripheral blood samples of recipients, their original transplant donor, and the third-party cell bank donor were assessed with the KMRtype Genotyping Primer/Probe Kit (GenDx, Utrecht, The Netherlands).
Statistical analysis
The cumulative incidence function was used to determine the cumulative CR rate to antiviral treatment and VST infusions at the end of the 6-month follow-up period, with death considered a competing risk. Twelve-month post-transplant cumulative incidence of relapse and nonrelapse mortality were also determined with this function. Overall survival was calculated by using the Kaplan-Meier method. A Spearman’s rank correlation was used to determine the significance of association between the AUC of the viral load and other variables. A 2-tailed Wilcoxon matched-pairs, signed-rank correlation was used to determine the significance of pre-infusion and maximum CMV-induced IFN-γ production within 100 days. For visualization of immune cell subsets over time, the fit of the trajectories for each population was performed in R,21 using a loess curve-fitting technique. Linear mixed models utilizing the lme4 package in R were employed to calculate the significance of the AUC of the viral load in each immune cell population.22
Results
Participant characteristics
Thirty-seven patients were recruited at 3 Australian centers from August 2017 through April 2020. Three patients did not receive treatment because there was no suitably HLA-matched product. All 3 patients expressed HLA-A11:01, to which responses were not routinely assessed after product manufacture. Two patients were excluded from the trial before VST infusion because of failure to satisfy eligibility criteria (for details see supplemental Methods 1). One patient died of septic shock and multiorgan failure after recruitment, but before the infusion. One pediatric patient was excluded retrospectively when it was realized that the patient was treated for viral reactivation 975 days after transplant and could not be assessed for trial end points. Of the 30 remaining patients, 27 were treated for CMV reactivation and 3 for EBV reactivation. Twenty-seven adult patients underwent transplant for hematological malignancies; 3 pediatric patients were treated for immune deficiency syndromes. Conditioning was myeloablative in 12 patients and reduced intensity in 18 patients. Most of the patients (22 of 30) underwent in vivo T-cell depletion. Seven of 27 CMV-seropositive recipients received transplants from CMV-seronegative donors. All patients were treated for viral infection within 7 days of initiation of antiviral therapy (median 4 days). For full participant characteristics, see Table 1.
Product characteristics
Forty-one VST infusions from 11 donors were administered during the trial, with products from 5 donors used in multiple patients (see Table 2). The most frequently shared HLA types were A2 and B7. VST products were mostly CD3+ (median, 97.9%; range, 96.2% to 99.4%), with the median percentage of CD3+CD4+ being 17.9% (range, 6.0% to 69.3%) and CD3+CD8+ 85.8% (range, 23.2% to 95.5%). Antigen-specific responses quantified by cytokine intracellular flow cytometry response to peptides of known HLA restriction are detailed in Table 2. Virus-specific MHC class I tetramers were available for 7 products and are detailed in Table 2.
Administration of VSTs
A total of 41 infusions were performed in 30 patients (1 infusion, n = 21; 2 infusions, n = 8; 4 infusions, n = 1). Viral load measurements in the period before initial VST infusion and at the time of initial viral detection are given in Table 1 and in supplemental Table 1 in the supplemental Material. Two patients received products from >1 donor. The median day of the first infusion was day 4 after initiation of antiviral treatment (range, 0-6 days) and day 55 after transplant (range, 19-83 days). All infusions were administered at a cell dose of 2 × 107/m2. The majority of the patients (23 of 30) were matched at 2 or more HLA antigens with the product at any class I locus, HLA DRB1 or HLA DQB1, whereas the remainder (7 of 30) were matched at a single locus. Antigen matches HLA C and DQB1 were counted toward the total degree of matching, but only matches at HLA A, B, and DRB1 were used to select a suitable product. Nine patients received multiple infusions for persistent viral replication. Patients 13 and 16 received products from >1 donor. Patient 13 received 4 infusions from 3 separate products and achieved a PR as best virological response. Patient 16 received 2 infusions of different products, both matched at HLA A2 with the transplant donor and patient. Both infusions resulted in a CR. The remaining 7 patients who received 2 infusions received a second infusion of the same product. The initial product used to treat patient 16 was not available at the time of the second VST infusion. See Table 2 for details.
Safety and toxicity of VSTs
There were 6 instances of adverse events (AEs) occurring within 24 hours of infusion, 4 of which were mild in severity (vomiting, altered taste sensation, fever, and 2 instances of hypertension). One child developed moderate hypertension after infusion that responded rapidly to diuretic treatment and an adult patient was admitted to the hospital with a fever and treated for Staphylococcus epidermidis sepsis. No AEs were attributed to VST infusion. All serious AEs (SAEs) are detailed in Table 3. Most of the of adverse reactions were related to infections. Non-VST–targeted viral and fungal infections are detailed in Table 4. Most of the patients (21 of 30) had infection with opportunistic pathogens not targeted by their VST infusion.
Virological response
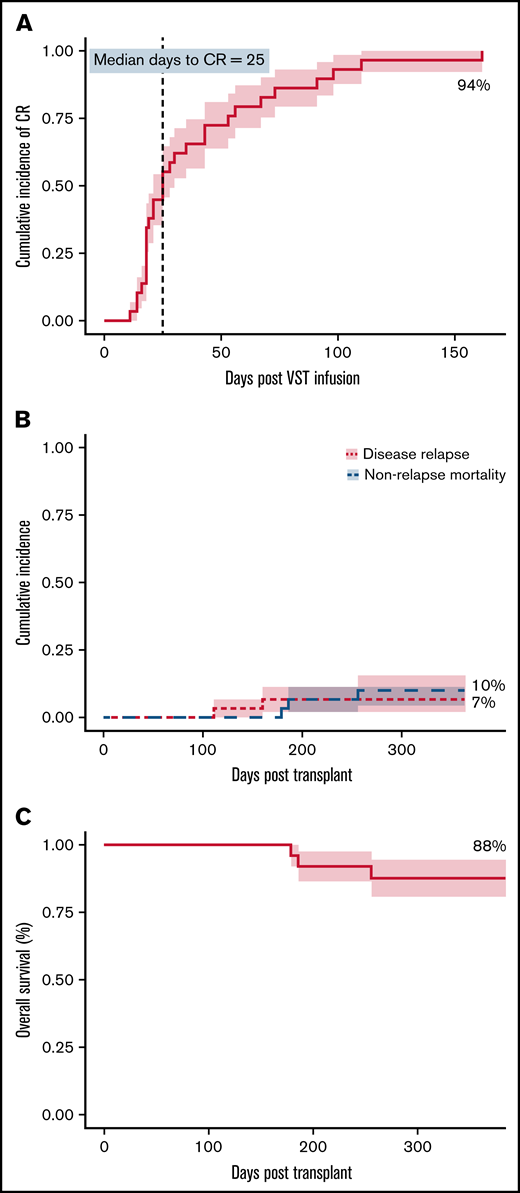
At 6 months, the cumulative incidence of virological CR was 94% (Figure 1A). Two of 30 patients achieved virological PR. No patients failed to respond. All 8 patients who received 2 infusions achieved a CR, whereas the single patient who received 4 infusions achieved a PR but subsequently died. There was a weak inverse correlation between HLA matching (1 vs 2 vs >2) and the number of antiviral courses, R2 = 0.1345; P = .046). The median day to CR was 25 days after the first VST infusion and was the same for patients who received only 1 VST infusion. Considering all 30 patients treated in the study, 20 (67%) had achieved their best virological response by day 42, and 24 (80%) by day 60 after initial infusion. Of the 28 patients achieving a CR, 17 (61%) and 22 (79%) achieved it by days 42 and 60, respectively. The median day to best virological response for all patients treated for CMV was 25 days and was unchanged in patients receiving antithymocyte globulin or in all patients receiving any type of in vivo or in vitro T-cell–depleted transplant. However, 7 CMV-seropositive patients with seronegative donors, including 1 of the 2 patients who achieved only partial virological control, had a median day to best virological response of 43 days.
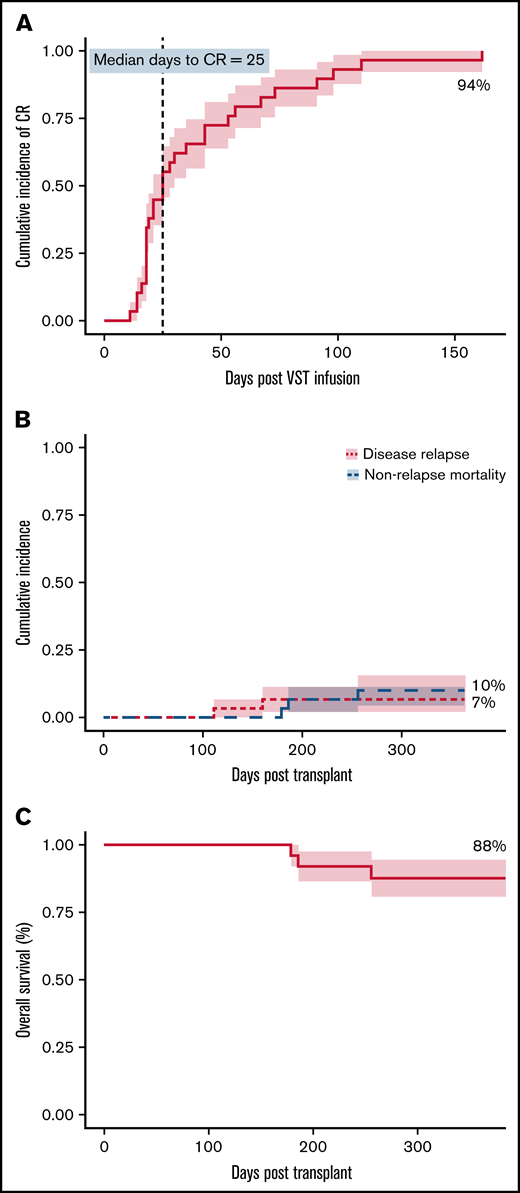
Clinical outcomes. (A) Cumulative incidence of complete response (CR) adjusted for competing risks, over the 6-month follow-up period after VST infusion. (B) Cumulative incidence of disease relapse and non-relapse mortality in the first 12 months after transplant. (C) Overall survival in the first 12 months after transplant. N = 30 for all.
Clinical outcomes. (A) Cumulative incidence of complete response (CR) adjusted for competing risks, over the 6-month follow-up period after VST infusion. (B) Cumulative incidence of disease relapse and non-relapse mortality in the first 12 months after transplant. (C) Overall survival in the first 12 months after transplant. N = 30 for all.
Of the 28 patients who achieved a CR, 23 remained virus PCR negative (n = 9) or below the level of quantitation (n = 14) for the duration of follow-up. Four patients had brief episodes of quantifiable reactivation but did not need additional VST or antiviral therapy after achieving CR. One patient had reactivation requiring a second infusion after the initial CR, and this patient remained PCR negative after the second CR.
Patients 13 and 26 achieved PRs. Patient 13 remained CMV PCR positive throughout a complicated post-transplant course and received 4 VST infusions in total, eventually dying of pulmonary veno-occlusive disease. Patient 26 had a recurrence of preexisting BK hemorrhagic cystitis after CMV VST infusion, resulting in rapid weaning of immunosuppression and the onset of refractory acute liver and gut GVHD, causing death.
We analyzed data from a historical control cohort of 78 patients who received initial treatment for CMV reactivation after allogeneic transplant and did not receive VSTs and compared their CMV-related outcomes to those of the 27 patients who received VSTs for CMV in this study. The post-transplant day of initial viral reactivation (29.5 vs 30.5 days) and day of initiation of antiviral treatment (48 vs 48 days) were similar in current and historical cohorts, respectively. The median duration of first antiviral course was 17.5 vs 23 days, respectively. A similar percentage of patients needed a second antiviral course (33.3% vs 31.1%), but a lower percentage of patients receiving VSTs needed a third course (7.4% vs 20.5%). The median total duration of CMV treatment in the first 12 months after transplant was 21.5 vs 32 days. The percentage of patients with CMV tissue disease at any time after transplant was 3.7% vs 20.5% for the VST-treated patients vs controls. Non-relapse mortality at 1 year was 11.1% vs 20.5% for VST-treated patients vs controls. However, caution must be exercised in comparing the groups, because a higher proportion of the control group underwent transplant with poor-risk disease (high or very high disease risk index, 11.1% vs 33.3% in the T-cell treated and control groups, respectively), and this may have contributed to the high rate of CMV tissue disease in the controls.
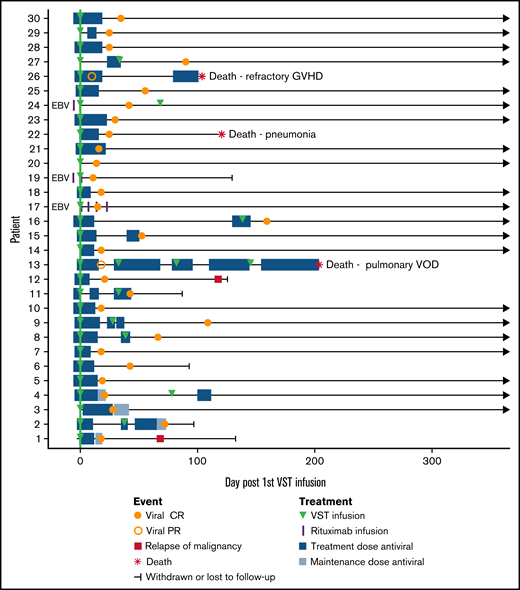
Three patients were treated for EBV infection (patients 17, 19, and 24): 2 for biopsy-proven post-transplant lymphoproliferative disorder (PTLD) and 1 for suspected PTLD on clinical presentation and imaging. All 3 patients achieved CR and sustained disease remission with a combination of EBV VSTs and rituximab. The median number of days receiving antiviral therapy for all patients was 4 before infusion (range, 0-6) and 18 after infusion (range, 0-155). Clinical outcomes are summarized in Table 5 and Figure 2.
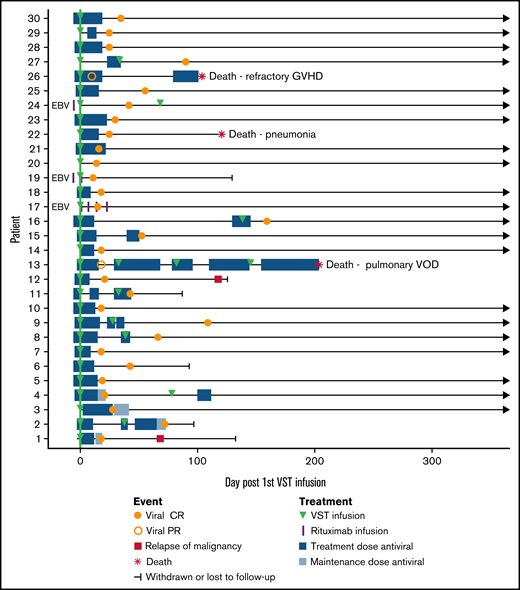
Composite swimmer plot showing the clinical course of patients over the 12 months after VST infusion. Patients appear in order of recruitment on the y-axis. Patients treated for EBV infection are noted (n = 3); all others were treated for CMV infection (n = 27). All patients received the first VST infusion at day 0 (green line), and their subsequent clinical courses are illustrated. Antiviral treatment is shown before and after infusion: blue boxes (CMV therapy) and purple vertical lines (rituximab doses for EBV).
Composite swimmer plot showing the clinical course of patients over the 12 months after VST infusion. Patients appear in order of recruitment on the y-axis. Patients treated for EBV infection are noted (n = 3); all others were treated for CMV infection (n = 27). All patients received the first VST infusion at day 0 (green line), and their subsequent clinical courses are illustrated. Antiviral treatment is shown before and after infusion: blue boxes (CMV therapy) and purple vertical lines (rituximab doses for EBV).
GVHD, relapse, and death
The overall rates of aGVHD (grades 2-4) and cGVHD after infusion were 13% (4 of 30) and 23% (7 of 30), respectively (Table 5). Two of 4 patients had aGVHD before VST infusion that had resolved sufficiently to enable infusion to proceed. One patient (patient 6) with previously resolved grade 3 (gut stage 3) aGVHD developed grade 4 (gut stage 4, liver stage 3, skin stage 3) GVHD 89 days after infusion during weaning from corticosteroids and was then enrolled in a trial of an experimental therapy for GVHD. The other patient (patient 16) did not have worsening of GVHD grade after VST. Two patients developed de novo acute GVHD after infusion. Patient 25 developed grade 3 aGVHD (stage 2 skin, stage 3 liver) 63 days after VST infusion, which subsequently resolved. Patient 26 developed de novo aGVHD of the liver (stage 3) and gut (stage 4) on day 57 after infusion in the context of rapid weaning of immunosuppression for management of severe BK hemorrhagic cystitis and died of GVHD. Of the 7 patients who developed cGVHD, 2 had severe disease (1 involving lung, 1 involving gut, liver, and skin) and 5 had mild/limited disease.
Overall survival at 1 year after transplant was 88%, with median survival not reached. Nonrelapse mortality at 1 year was 10%, and cumulative incidence of relapse was 7% (Figure 1B-C). Median follow-up was 385 days after the first VST infusion (range, 88-1029). Three of 30 patients died within 1 year of transplant; 1 of respiratory failure caused by pulmonary veno-occlusive disease, 1 of refractory GVHD, and 1 of pneumonia. One of 30 patients died more than 1 year after transplant (day 499), from EBV PTLD (this patient had been treated for CMV infection and achieved a CR). Patient outcomes are summarized in Table 5 and Figure 2.
Performance status
Excellent performance status was maintained in VST recipients. Twenty-five of 27 alive at 12 months (or at last follow-up if <12 months) had an ECOG status of 0 or 1, 1 patient had a status of 2, and 1 had a status of 3 (Table 3). The median duration of hospital inpatient stay after infusion was 6.5 days (range, 0-43). Of the 30 patients, 13 had no inpatient days in the 6 months after the first VST infusion, 10 had <3 weeks, 2 had from 3 to 6 weeks, and 5 had >6 weeks. Details of clinical course and post-infusion hospital admissions are described in Table 5.
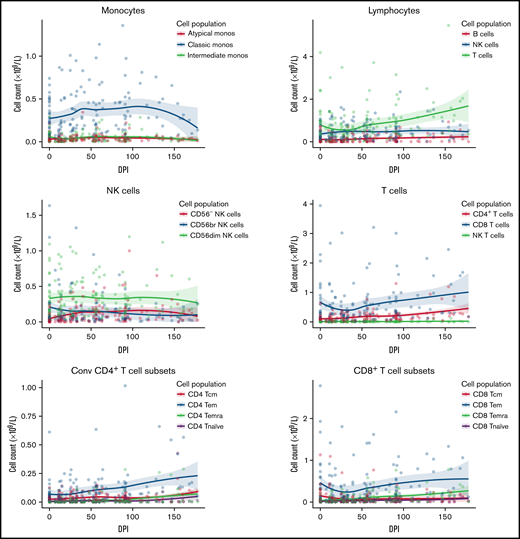
Immune reconstitution
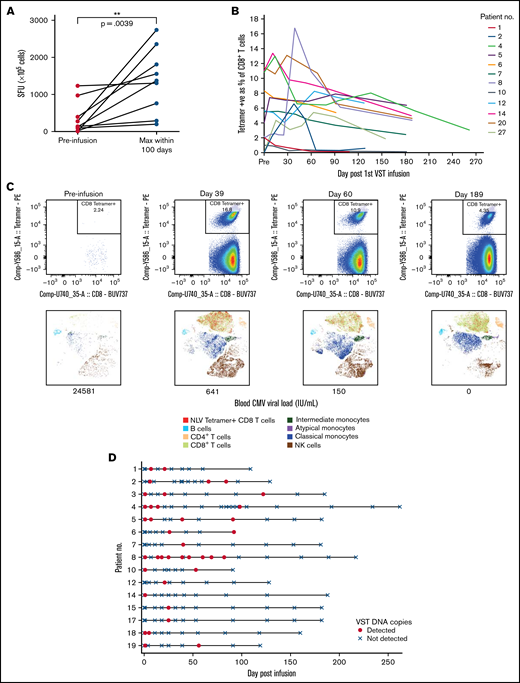
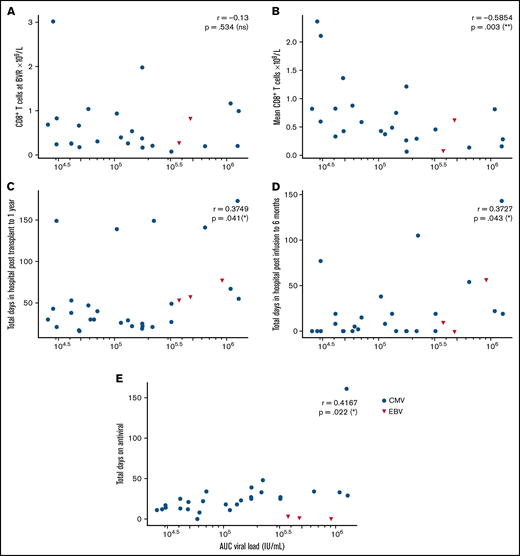
Immune reconstitution after VST infusion was assessed with a 20-marker flow cytometry panel in 25 of 30 patients. Results are shown in Figures 3 and 4 and in supplemental Figures 1 to 4. After an initial decline, CD8+ T-cell expansion was observed over time (median CD8+ preinfusion count, 0.31 × 109/L; 100 days after infusion, 0.72 × 109/L). The predominant subset within both CD8+ and CD4+ populations was CD45RA−CD62L− effector memory (em) cells, with a steady increase over time in CD8+CD45RA+CD62L− terminally differentiated effector memory (emra) cells and a later increase in the analogous CD4+ population. Expression of exhaustion marker PD-1 and activation marker HLA-DR on T-cells was highest before infusion and decreased in the first 50 days after infusion (supplemental Figure 1). The progress of other leukocyte populations after VST infusion is shown in supplemental Figure 1. A significant inverse correlation was found between mean CD8+ count after infusion and the AUC of viral load, such that patients with higher CD8+ counts had lower viral AUCs over the period of follow-up (Figure 5B; r = −0.5854; P = .002). AUC correlated positively with total days in the hospital in the first year after transplant and with total days in the hospital in the first 6 months after VST infusion, as well as with total days receiving antiviral treatment (Figure 5C-E). There was no correlation between AUC and CD8+ T-cell numbers at the time of best viral response (Figure 5A). Other leukocyte subsets that correlated inversely with AUC included total lymphocyte count; total T, NKT, and CD4+ em cells; CD8+ HLA-DR+, CD8+ CD86+, and CD8+ naïve cells; CD8+ em cells; and CD8+ emra cells (supplemental Figure 2).

Immune reconstitution of 25 patients by flow cytometry, showing major cell populations and their subsets over time. Median T-cell subsets are shown, with the predominant CD4+ and CD8+ subset being CD45RA-CD62L- effector memory cells. Additional subsets are visualized in supplemental Figure 1. DPI, day post 1st VST infusion; br, bright; cm, central memory; Conv, conventional; em, effector memory; emra, terminal effector memory.
Immune reconstitution of 25 patients by flow cytometry, showing major cell populations and their subsets over time. Median T-cell subsets are shown, with the predominant CD4+ and CD8+ subset being CD45RA-CD62L- effector memory cells. Additional subsets are visualized in supplemental Figure 1. DPI, day post 1st VST infusion; br, bright; cm, central memory; Conv, conventional; em, effector memory; emra, terminal effector memory.
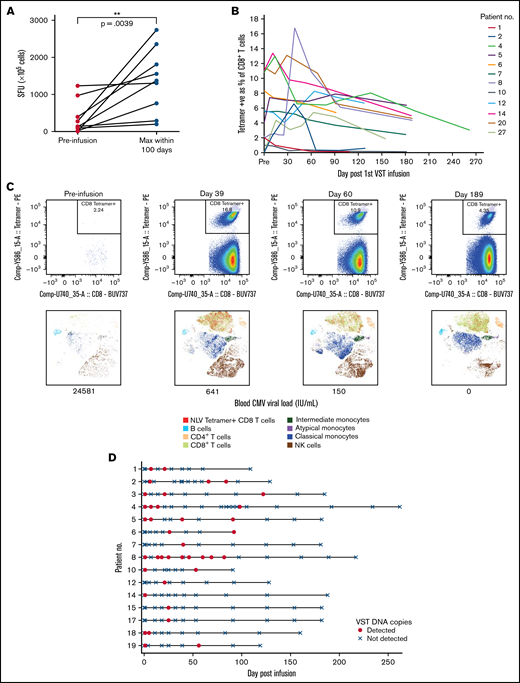
Virus-specific T-cell responses and cell persistence in post-infusion samples. (A) ELISpot assay showing preinfusion and maximum CMV-induced IFN-γ production within 100 days after infusion. Statistical significance was evaluated with a 2-tailed Wilcoxon matched-pairs signed-rank test. (B) Tetramer responses for 12 patients with >1% expression, showing elevation and persistence over time. (C) Tetramer responses for patient 8 shown in composite at 4 time points. For each time point, biaxial flow plots, tSNE of major cell populations, and blood viral load (below each tSNE plot) are shown. (D) Microchimerism assessed by ddPCR in 15 patients. Informative insertion-deletion polymorphisms (indels) that distinguished the third-party VST donor from the transplant donor and recipient were identified. Adoptively transferred VST product was detected in the peripheral blood of all patients up to day 120 after infusion, all quantified below 3 copies per 100 ng of DNA. Day 0 results were from samples taken 2 hours after VST administration. Patient 6 withdrew from the study and had no further time points after day 92. tSNE, t-distributed stochastic neighbor embedding.
Virus-specific T-cell responses and cell persistence in post-infusion samples. (A) ELISpot assay showing preinfusion and maximum CMV-induced IFN-γ production within 100 days after infusion. Statistical significance was evaluated with a 2-tailed Wilcoxon matched-pairs signed-rank test. (B) Tetramer responses for 12 patients with >1% expression, showing elevation and persistence over time. (C) Tetramer responses for patient 8 shown in composite at 4 time points. For each time point, biaxial flow plots, tSNE of major cell populations, and blood viral load (below each tSNE plot) are shown. (D) Microchimerism assessed by ddPCR in 15 patients. Informative insertion-deletion polymorphisms (indels) that distinguished the third-party VST donor from the transplant donor and recipient were identified. Adoptively transferred VST product was detected in the peripheral blood of all patients up to day 120 after infusion, all quantified below 3 copies per 100 ng of DNA. Day 0 results were from samples taken 2 hours after VST administration. Patient 6 withdrew from the study and had no further time points after day 92. tSNE, t-distributed stochastic neighbor embedding.
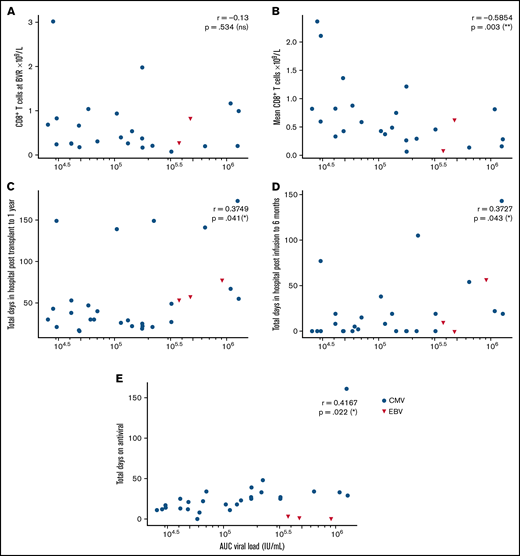
Correlations between AUC of the viral load and various clinical and immunological outcomes, by Spearman rank correlation coefficient. (A) Nonsignificant correlation with CD8+ T-cell count at the time of best viral response (n = 25). (B) Significant negative correlation with mean CD8+ T-cells across the follow-up period (n = 25). (C) Significant positive correlation with total days in hospital from transplant to 1 year later (n = 30). (D) Significant positive correlation with total days in hospital from first VST infusion to 6 months after infusion (n = 30). (E) Nonsignificant correlation with total days of antiviral treatment (n = 30).
Correlations between AUC of the viral load and various clinical and immunological outcomes, by Spearman rank correlation coefficient. (A) Nonsignificant correlation with CD8+ T-cell count at the time of best viral response (n = 25). (B) Significant negative correlation with mean CD8+ T-cells across the follow-up period (n = 25). (C) Significant positive correlation with total days in hospital from transplant to 1 year later (n = 30). (D) Significant positive correlation with total days in hospital from first VST infusion to 6 months after infusion (n = 30). (E) Nonsignificant correlation with total days of antiviral treatment (n = 30).
Elispot demonstrated that CMV-induced IFN-γ production by lymphocytes increased over time after infusion (Figures 4A-B). Nineteen patients were tested with HLA tetramers for CMV or EBV epitopes (supplemental Figure 3). In some patients, tetramer-positive cells were present before VST infusion. These may have been recipient in origin, from incomplete donor T-cell chimerism or from transfer of antigen-specific T-cells from the stem cell donor product. Despite the presence of tetramer-positive cells, all patients satisfied criteria for initiation of antiviral treatment. Twelve of 19 patients showed a rapid increase in virus-specific T-cells to between 1% and 13% of CD8+ T-cells, and the cells persisted 6 months after infusion (Figure 4B-C). Expression of CD57 on tetramer-positive cells rose quickly, 1 to 2 weeks after VST administration, and remained elevated at 6 months after infusion. Expression of HLA-DR, PD-1, and Tim-3 decreased over the same period. In all of those patients, the presence and persistence of virus-specific T-cells corresponded with virological CR. Patient 13, who achieved only a PR, showed poor recovery of adaptive immunity, with dominance of innate effectors (supplemental Figure 4).
Microchimerism assessed by droplet digital PCR (ddPCR) was used to determine the persistence of the third-party VST product in 15 patients. Although adoptively transferred VSTs were detected at least once in all 15 patients up to 120 days after infusion (median duration, 55 days; Figure 4D), in all cases, detection was below the linear range of the assay that would allow for quantitation (0.032% or ≤3 gene copies per 100 ng of DNA). Tetramer-sorting of cells before microchimerism testing did not detect the persistence of third-party cells beyond 120 days.
Discussion
Partially HLA-matched third-party VSTs issued from a bank of cryopreserved cells offer an alternative to fully HLA-matched, donor-derived VSTs for post-transplant viral infections. They can be available on short notice, do not require pre-transplant manufacture from the stem cell donor, and have clinical benefit in the short11,23,24 and long term.9,12 Until now, third-party VSTs have been administered only in the setting of resistant or relapsing infection. In our study, we assessed the safety and effect of administering partially HLA-matched, third-party VSTs within 7 days of initiating standard-of-care antiviral treatment for first post-transplant CMV or EBV viral reactivation. Patients commenced antivirals according to widely accepted clinical criteria for treatment initiation and were observed for infusion-related toxicities, immune reconstitution, and clinical course. Infusion reactions in the first 24 hours were mild and infrequent. Rates of GVHD (acute and chronic) and SAEs during the follow-up period were within expectations for the studied population of postallogeneic transplant patients. No SAEs were attributable to the VST infusion. Overall, third-party VSTs administered early in the course of treating viral infection in this way are safe and without significant toxicity.
The overall virological CR rate for combined antiviral and third-party T-cell therapy was 94%, and the remaining patients achieved a PR. The combination of VSTs and conventional antiviral treatment led to a high rate of sustained viral clearance. Only 3 patients needed retreatment with VSTs for a reactivation episode after initial CR in the follow-up period, and all 3 achieved sustained virological control after retreatment. Outcomes in the entire cohort were excellent, with overall survival at 1 year after transplant of 88%, nonrelapse mortality of 10%, and relapse at 1 year of 7%. No patients died of the infection for which they were treated, and the causes of death were not attributable to VST infusion.
Virological clearance coincided with T-cell recovery, with CD45RA−CD62L− terminal effector cells the predominant subset in both CD8+ and CD4+ T-cell populations. Given the early time point of third-party T-cell administration and the brevity and degree of their persistence by ddPCR, it is likely that recovery of these cells reflected donor-derived immunity or, in the case of reduced intensity conditioning, temporary persistence of host immunity. Gradual expansion of CD8+CD45RA+CD62L− terminally differentiated effector memory cells also occurred over time. We noted evidence of viral immunity before administration of VSTs that may represent residual host T-cell function or cells transfused with the stem cell donor product. Either way, the immunity present before infusion was insufficient to control post-transplant viremia. Post-infusion immunity was unlikely to derive directly from expansion of the infused third-party T-cells based on the result of ddPCR assessment of chimerism and may have come from augmented stem cell donor and/or residual host immune function. The magnitude of T-cell subset expansion after VST infusion was not as great as we found in a previous trial of refractory viral infections,12 possibly because the patients in this trial were treated soon after transplant and earlier in the course of their infection episode. The elevated expression of HLA-DR and PD-1 on T-cells before VST infusion is consistent with an inflammatory state associated with active viral infection.25 Expression of both decreased slowly after VST infusion. Only 2 patients failed to achieve virological CR. In the single patient with samples available for analysis, there was a failure of T-cell recovery over all subsets and a failure of differentiation to CD45RA+CD62L− terminal effector memory phenotype (supplemental Figure 4).25
In patients who achieved virological control, we detected tetramer-positive, viral antigen–specific T-cell populations and observed enhanced IFN-γ production after viral antigen stimulation, changes that persisted to the end of the follow-up period. DNA from adoptively transferred VST product was detected only in post-infusion samples up to 120 days after infusion and at very low levels, suggesting that the VST response may be at least partially explained by induction of an endogenous T-cell population with antiviral capacity.
Opportunistic coinfections with other viruses and fungi were present in most of the patients in the trial and most frequently involved infection with CMV and EBV in the same patient (Table 4). The correlation between multiple infections and increased posttransplant mortality is well recognized.1 In patients with multiple posttransplant infections, it is possible that fundamental defects in immune recovery predispose to multiple infections and recurrence of malignancy. Our data demonstrate the safety and efficacy of early administration of third-party VSTs, together with antiviral therapy in controlling viral infection after transplant, but suggest that targeting more than 1 pathogen may be required for the greatest benefit. Third-party VSTs given early after initiation of antiviral treatment are associated with specific immune reconstitution and excellent viral control, as well as with low rates of nonrelapse mortality and disease recurrence. The feasibility of the approach is excellent, because VSTs from a third-party bank can be made available within a few days of request across multiple national participating centers. Although our study is limited by its small sample size and the shortcomings of a nonrandomized trial, the outcomes suggest that a larger study examining the effects of third-party VSTs is warranted.
Acknowledgments
The study was supported by grants from the Leukemia and Lymphoma Society of the United States (CCR-16-100) and the Rising Tide Foundation for Clinical Cancer Research (RTF6001-17). Grant assistance was provided by the Cancer Institute of NSW and Cancer Council NSW. W.J. was supported by a scholarship from the Haematology Society of Australia and New Zealand and the Leukaemia Foundation. E.B. was a Cancer Institute NSW Postgraduate Fellow and recipient of a NSW Ministry of Health Early/Mid-Career Cell and Gene Therapy Fellowship. Microchimerism assays, DNA quantification, and flow cytometry were performed at the Westmead Scientific Platforms, which are supported by the Westmead Research Hub, the Westmead Institute for Medical Research the Cancer Institute of New South Wales, the National Health and Medical Research Council, and the Ian Potter Foundation.
Authorship
Contribution: W.J. supervised the clinical trial, recruited patients, performed correlative experiments, analyzed data, and prepared the manuscript; L.E.C. supervised cell manufacture; S.A. performed and supervised cell manufacture; G.S. recruited patients and assisted with trial management; J.S. and R.S. performed correlative experiments; H.M.M. advised on flow cytometry; E.P., A.S.C., and N.M. analyzed data; G.M. collected and analyzed data; K.P.M. and V.A. supervised product distribution; A.G.S., D.R., C.M.B., and P.J.S. recruited patients; E.B. recruited and treated patients, supervised correlative experiments, analyzed data, and assisted with manuscript preparation; and D.J.G. conceived the study, obtained the funding, directed the trial, and wrote the manuscript.
Conflict-of-interest disclosure: L.E.C., K.M., E.B., and D.J.G hold patents in adoptive cell therapy for opportunistic infection and malignancy. E.B. holds advisory board membership with AbbVie, Astellas, MSD, Novartis, BMS, Bastion Education and Atara Biotherapeutics. E.B. reports research funding from MSD. The remaining authors declare no competing financial interests.
Correspondence: David J. Gottlieb, Department of Medicine, Westmead Hospital, Corner Hawkesbury Rd and Darcy Rd, Westmead, NSW 2145, Australia; e-mail: david.gottlieb@sydney.edu.au.