

Key Points
WGS analysis shows complex structural variants of chromosome 2, and duplication of 22q11.23 in one patient with TEMPI syndrome.
Duplication of 22q11.23 in plasma cells may result in elevated level of MIF, which may contribute to the pathophysiology of TEMPI syndrome.
TEMPI syndrome (telangiectasias, elevated erythropoietin level and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting) is a newly defined multisystemic disease with its pathophysiology largely unknown. Here, we report the whole-genome sequencing (WGS) analysis on the tumor-normal paired cells from a patient with TEMPI syndrome. WGS revealed somatic nonsynonymous single-nucleotide variants, including SLC7A8, NRP2, and AQP7. Complex structural variants of chromosome 2 were found, particularly within regions where some putative oncogenes reside. Of potential clinical relevance, duplication of 22q11.23 was identified, and the expression of the located gene macrophage migration inhibitory factor (MIF) was significantly upregulated in 3 patients with TEMPI syndrome. Importantly, the level of serum MIF in one patient with TEMPI syndrome was significantly decreased in accordance with the downtrend of plasma cells, M-protein, hemoglobin, and erythropoietin and the improvement of telangiectasias, perinephric fluid collections, and intrapulmonary shunting after treatment with plasma cell–directed therapy. In conclusion, our study provides insights into the genomic landscape and suggests a role of MIF in the pathophysiology of TEMPI syndrome.
Introduction
TEMPI syndrome is a rare multisystemic disease characterized by the pentad of telangiectasias, elevated erythropoietin level and erythrocytosis, monoclonal gammopathy, perinephric fluid collections,and intrapulmonary shunting.1 It is hypothesized to be a result of monoclonal plasma cells (PCs), aspatients show dramatic response to PC-directed therapy.2 However, the pathophysiology of TEMPI syndromeremains elusive, and little is known about its genomic changes compared with other PCneoplasms.3 In this study, we performed whole-genome sequencing (WGS) analysis on monoclonalPCs and germline control from a patient with TEMPI syndrome to study genomic changes ofTEMPI syndrome and further identified a possible role of macrophage migration inhibitoryfactor (MIF) in the pathophysiology of TEMPI syndrome in 3 patients.
Methods
Patients and samples
Bone marrow (BM) aspirates and serum samples of patients with TEMPI syndrome and healthy donors, and serum samples of patients with multiple myeloma were obtained after written informed consent was obtained. Purified tumor cells from 3 patients with TEMPI syndrome and primary PCs from 4 healthy donors were obtained from BM aspirates using CD138+ magnetic beads selection (Miltenyi Biotec, Auburn, CA) after isolation of mononuclear cells using Ficoll density gradient centrifugation according to the recommended protocols. PCs obtained from patient 1 were divided into 2 parts; one was used for WGS and the other one was used for quantitative polymerase chain reaction(PCR). However, given the limited number of PCs obtained frompatient 2 and patient 3, the cells were only used for quantitative real time-PCR. Buccal cells obtained from patient 1 were used as germline control.
Samples for WGS were preserved as dry pellets at −80°C. The genomic DNA from the preserved samples was extracted with theTIANamp Genomic DNA Kit (Tiangen) following the manufacturer’s instructions. The quality and quantity of the DNA were assessed with the use of NanoPhotometer (Implen, Westlake Village, CA) and Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA), respectively, and both were accepted for further analysis. These studies were conducted in accordance with the Declaration of Helsinki, and studies were approved by the institutional review board of Huazhong University of Science and Technology.
WGS and data analysis
A volume of 1 μg of genomic DNA was used for the preparation of paired-end libraries according to the TruSeq DNA Sample Preparation Guide (Illumina, 15026486 Rev. C). Briefly, DNA was fragmented using the Covaris E210 sonicator (Woburn, MA). The ends of the DNA fragments were repaired and phosphorylated followed by 3' end adenylation. Paired-end DNA adapters were ligated, and the resulting constructs size selected for ∼500-bp fragments. The adapter ligated fragments from the excised gel band were then purified using the MinElute Gel Extraction Kit and enriched with 12 cycles of PCR. The concentration and size distribution of the libraries were determined on an Agilent Technologies 2100 Bioanalyzer using DNA 1000 chip. Libraries were quantified using a Bio-Rad KIT iQ SYBR GRN on Bio-Rad CFX 96 according to Illumina’s library quantification protocol. The library was loaded onto paired end-flow cells and sequenced using 150-bp paired-end reads on the illumina HiSeq 2000 platform (Illunima).
Mean sequencing depth was ×60 for the samples with an average of 99.5% of reads mapping to the hg19 reference (supplemental Table 1). Reads mapping and filtering were performed using the BWA, SAMtools, and GATK software. Somatic single-nucleotide variants (SNVs) and insertions or deletions were called and filtered using MuTect2, and then were annotated by ANNOVAR using the RefGene, tfbsConsSites, 1000 Genome, COSMIC, dbSNP, ClinVar, and dbNSFP reference databases. If an SNV or insertions or deletions were in a low-quality region or germline mutation, it was discarded. Somatic structural variants (SVs) were analyzed using DELLY2, and the results were annotated by ANNOVAR. An SV that was not found in germline control cells was defined as somatic SV.
Quantitative real-time amplification of messenger RNAs
Total RNAwas isolated using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Complementary DNA was synthesized with PrimeScript RT reagent Kit (Takara, Dalian, China), and then quantitative real time–PCR was performed using SYBR Green real time-PCR Kit (Takara). The primers for MIF were as follows: forward: 5'-ACCGCTCCTACAGCAAGC-3', reverse: 5'-CGCGTTCATGTCGTAATAGTTG-3'. Glyceraldehyde-3-phosphate dehydrogenase was used as an internal control, and the primers were as follows: forward: 5'-GGTGAAGGTCGGAGTCAACGG-3', reverse: 5'-CCTGGAAGATGGTGATGGGATT-3'. All PCR reactions were carried out in triplicate on ABI 7500 FAST Real Time PCR System (Applied Biosystems), and relative expression was evaluated by the comparative 2−ΔΔCT method.
MIF measurement
MIF levels in serum and BM aspirates from patients with TEMPI syndrome and healthy individuals, and in serum from 116 patients with multiple myeloma were determined by flow cytometry using a bead-based premixed multiplex kit/Human Custom 9-plex Kit (PN: T1C097809) (AimPlex Biosciences) following the recommended protocols. The reference range of serum MIF in healthy individuals was set up according to the manufacturer’s instructions.
Results and discussion
A 52-year-old woman (patient 1) was referred to our hospital for evaluation of unexplained erythrocytosis and proteinuria for 5 years in July 2017. On the day of her clinic visit, cyanosis and skin telangiectasias were on her hands, arms, face, and trunk (Figure 1A-B). Her hemoglobin was 190 g/L, and serum erythropoietin was 106 IU/L (reference range: 4.3 to 29 IU/L). BCR/ABL translocation and mutations of JAK2V617F, JAK2 exon 12, CALR, and MFL were negative. BM aspirate showed PCs making up 7% of the marrow cellularity, which were monoclonal with an immunophenotype that was CD19−, CD20−, CD56+, CD38+, CD138+, ckappa+ (supplemental Figure 1). Serum protein electrophoresis and immunofixation analysis showed 7.1 g/L immunoglobulin G-κ monoclonal protein. Arterial blood gas revealed pCO2 27.4 mmHg (normal range: 35 to 45 mmHg), pO2 57.9 mmHg (normal range: 80 to 100 mmHg), and alveolar-arterial oxygen gradient 56.9 mmHg (normal range: <30 mmHg). Computed tomography showed right-sided perinephric fluid collection (Figure 1C). Lung perfusion scintigraphy showed a presence of 21.3% intrapulmonary shunting (Figure 1D). The patient was diagnosed with TEMPI syndrome and started on a combined chemotherapy of TCD (thalidomide 100 mg, cyclophosphamide 300 mg/m2, and dexamethasone 40 mg). After 2 cycles, she had a normalized erythropoietin level and slight decrease of hemoglobin and serum M-protein (Figure 1E). However, her telangiectasias and hypoxemia did not improve. Her right-sided perinephric fluid collection disappeared, while a large amount fluid collection developed around the left kidney (Figure 1C). She was then started on a VCD regimen (bortezomib 1.3 mg/m2, cyclophosphamide 200 mg/m2, and dexamethasone 20 mg). After 3 cycles of therapy, she had dramatic clinical improvement with disappearance of the cyanosis, telangiectasias, and perinephric-fluid collection, normalized erythrocyte count and erythropoietin level, and significantly ameliorated intrapulmonary shunting (Figure 1A-E). Flow cytometry of the BM was negative for phenotypically aberrant clonal PCs (supplemental Figure 2), and the serum M-protein was reduced to 1.0 g/L. After 6 cycles, all her symptoms before admission disappeared, and the serum M-protein was decreased to 0.8 g/L.
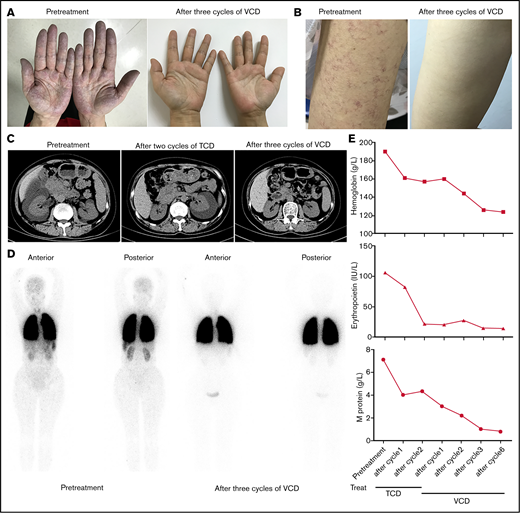
Clinical response of patient to treatment with TCD or VCD regimen. (A, B) The disappearance of cyanosis from the patient’s hands (A) and telangiectasias from her arm (B) after treatment with 3 cycles of VCD regimen. (C) Computed tomography scans of abdomen showing resolution of perinephric fluid collections after treatment with TCD and VCD regimen. (D) 99mTc-labeled macroaggregated albumin lung perfusion scintigraphy showing the decrease of intrapulmonary shunting after treatment with 3 cycles of VCD regimen (right side). The presence of intrapulmonary shunting was evidenced by the tracer uptake over the brain, liver, spleen, and kidneys in addition to the lungs. (E) Trend of hemoglobin (upper), erythropoietin (middle), and serum M-protein levels (lower) after treatment with TCD and VCD regimen.
Clinical response of patient to treatment with TCD or VCD regimen. (A, B) The disappearance of cyanosis from the patient’s hands (A) and telangiectasias from her arm (B) after treatment with 3 cycles of VCD regimen. (C) Computed tomography scans of abdomen showing resolution of perinephric fluid collections after treatment with TCD and VCD regimen. (D) 99mTc-labeled macroaggregated albumin lung perfusion scintigraphy showing the decrease of intrapulmonary shunting after treatment with 3 cycles of VCD regimen (right side). The presence of intrapulmonary shunting was evidenced by the tracer uptake over the brain, liver, spleen, and kidneys in addition to the lungs. (E) Trend of hemoglobin (upper), erythropoietin (middle), and serum M-protein levels (lower) after treatment with TCD and VCD regimen.
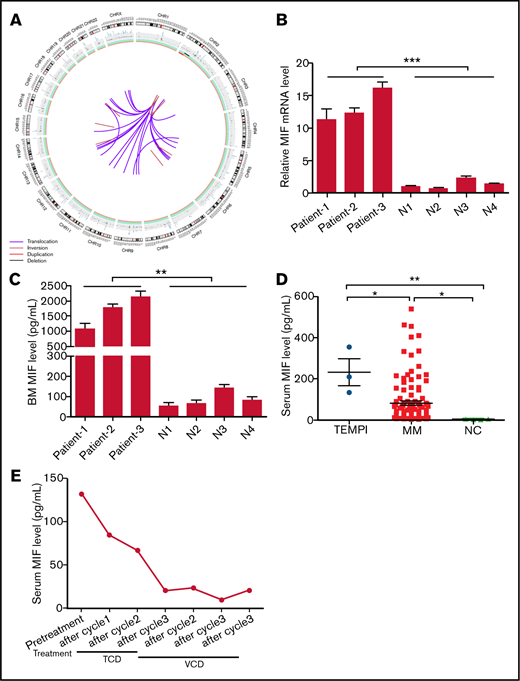
Circos plot of somatic SVs and the expression of MIF in patients with TEMPI syndrome. (A) The center, color lines indicate the presence of translocations and inversions. In the middle ring, the red and black lines indicate presence of duplication and deletion, respectively. The outermost ring indicates chromosome number. (B-D) Expression level of MIF mRNA in CD138+ PCs (B), and secreted MIF protein in BM aspirates (C) from healthy individuals (N) and 3 patients with TEMPI syndrome. (D) The level of serum MIF in healthy individuals (NC) and patients with TEMPI syndrome and multiple myeloma (MM; n = 116). (E) Serum level of MIF in the patient 1 after treatment with TCD and VCD regimen. Statistical analyses were performed using the unpaired Student t test. *P < .05; **P < .01; ***P < .001.
Circos plot of somatic SVs and the expression of MIF in patients with TEMPI syndrome. (A) The center, color lines indicate the presence of translocations and inversions. In the middle ring, the red and black lines indicate presence of duplication and deletion, respectively. The outermost ring indicates chromosome number. (B-D) Expression level of MIF mRNA in CD138+ PCs (B), and secreted MIF protein in BM aspirates (C) from healthy individuals (N) and 3 patients with TEMPI syndrome. (D) The level of serum MIF in healthy individuals (NC) and patients with TEMPI syndrome and multiple myeloma (MM; n = 116). (E) Serum level of MIF in the patient 1 after treatment with TCD and VCD regimen. Statistical analyses were performed using the unpaired Student t test. *P < .05; **P < .01; ***P < .001.
To study the possible molecular basis of TEMPI syndrome, paired-end WGS was conducted to analyze the genomes of the monoclonal PCs and germline control from the patient. We identified 25 somatic exonic SNVs. Among the SNVs, 18 were nonsynonymous and located in 17 genes (supplemental Table 2). Of the 17 genes, SLC7A8, POTEE, NRP2, AQP7, and PLCD3 were previously reported as tumor-associated genes. Interestingly, the SNV located in SLC7A8 was predicted as a damaging mutation by both PolyPhen-223 and SIFT24 software, and NRP2 and AQP7 were 2 of the somatically mutated genes identified in the Multiple Myeloma Research Consortium cohort of 38 myeloma genomes.4
SVs analysis revealed the presence of 38 somatic SVs (Figure 2A; supplemental Tables 3 and 4). Of interest, complex SVs occurred in chromosome 2, including depletion of 2p15-p22.3, duplication of 2p22.3-p25.3, and inversions of 2p22.3-p24.3, 2p22.3-q14.3, and 2q21.1 (supplemental Table 3). All of these changes occurred in regions where many important tumor-associated genes reside, such as MYCN (2p24.3), REL (2p16.1), BCL11A (2p16.1), BCL2L11 (2q13), MAPK4 (2p22.1), and TP53I3 (2p23.3). In addition, of all 24 translocations, 22 were associated with chromosome 2, and all the translocation breakpoints in chromosome 2 were located in the region of 2p22.3 (supplemental Tables 3 and 4). SVs of chromosomes 2, including gains at 2p25.3-p22.3, 2p22.3, and 2p16.2-p14, frequently occurred in chronic lymphocytic leukemia patients and were significantly related to poor prognosis.5 Therefore, further investigation would be needed to examine the significance of complex SVs on chromosome 2 in TEMPI syndrome.
Of potential clinical relevance, SVs analysis showed the duplication of 22q11.23, where the gene of MIF is located, in monoclonal PCs (supplemental Table 3). Compared with healthy controls, the level of MIF messenger RNA (mRNA) in CD138+ PCs and secreted MIF protein in BM from the patient was markedly upregulated (Figure 2B-C). In accordance, elevated level of serum MIF was found in the patient, which was ∼ 16.5-fold higher than the upper limit of normal range (reference range: 3.99 to 7.93 pg/mL) (Figure 2D). The upregulation of MIF mRNA in CD138+ PCs (Figure 2B), and the secreted MIF protein in BM (Figure 2C) was further confirmed in another 2 patients (patient 2 and patient 3; clinical information of the 2 patients is shown in supplemental Table 5) with TEMPI syndrome. Consistent with previous study,6 the level of serum MIF in patients with MM (82.10 ± 10.15 pg/mL) was higher than that in healthy controls (3.977 ± 0.684 pg/mL) (Figure 2D); however, it was lower than that in patients with TEMPI syndrome (231.7 ± 64.89 pg/mL; P = .0198). These results indicate that the duplication of 22q11.23 may result in the upregulation of MIF in monoclonal PCs, which will further increase the secretion of MIF into BM and serum.
MIF is a pleiotropic cytokine with proinflammatory, chemokine-like, and proangiogenic properties.7 Wang et al8 have reported that MIF plays an important role in retinal neovascularization by inducing the expression of vascular endothelial growth factor and erythropoietin, and Oda et al9 have found that MIF could induce both the expression levels and the transcriptional activity of hypoxia-inducible factor-1a, a crucial transcription factor for erythropoietin production. Arteriovenous malformations and vascular dilation are 2 common causes of intrapulmonary shunting and telangiectasia. Previous studies have established that MIF was involved in the pathogenesis of arteriovenous malformations and could induce the production of nitric oxide, a potent vasodilator, through upregulating the expression of inducible nitric oxide synthase.10-12 In addition, the upregulation of MIF may promote the formation of perinephric fluid collections because of the finding that MIF is an important promotor of peritoneal fluid and pleural effusion.13,14 Of note, we found all the 3 patients had the clinical characteristics of peritoneal fluid and pleural effusion (supplemental Table 5), and previous studies also showed that 5 patients with TEMPI syndrome exhibited peritoneal fluid or pleural effusion.15-19 In this study, we also found that the level of serum MIF in patient 1 was significantly decreased after treatment with the VCD regimen, which was changed in accordance with the downtrend of PCs, M-protein, hemoglobin, erythropoietin, and the improvement of telangiectasias, perinephric fluid collections, and intrapulmonary shunting (Figure 2E). These studies revealed a possible role of MIF in the development of intrapulmonary shunting, telangiectasias, elevated erythropoietin levels, and perinephric fluid collections, and furthermore, with the association between MIF levels and response to treatment in our patient, supporting that the duplication of 22q11.23 and upregulation of MIF in the monoclonal PCs may be involved in the pathophysiology of TEMPI syndrome.
Gene mutations, such as KRAS, NRAS, DIS3, and LTB, and chromosomal abnormalities including IGH (14q32) translocations, RB1 (13q14) deletion, 1q gain, hyperdiploidy, and 17p deletion, were observed in MM and monoclonal gammopathy of undeterminedsignificance.20 However, these reported mutations were not found in our patient, indicating that although TEMPI syndrome was considered a PC dyscrasia, significant molecular genetic differences might also exist between TEMPI syndrome and other PC malignancies. In summary, our study provides insights into the genomic landscape and suggests a role of MIF in the pathophysiology of TEMPI syndrome.
Acknowledgments
The authors thank all members of the study team, the patients, and healthy donors.
This work was supported by the National Natural Science Foundation of China, 81670197 and 81974007 (C.S.), 82000223 (J.X.), Clinical Research Physician Program of Tongji Medical College, HUST (C.S.).
Authorship
Contribution: C.S. and Y.H. conceived the premise of the work; J.X., Y.H., and C.S. conceived the methodology; J.X. and C.S. performed the investigation; J.X., J.L., H.H., A.X., and H.Y. provided the formal analysis; J.L., L.L., B.Z., H.H., D.H., A.X., L.C., B.Z., andF.Z. provided the resources; and J.X., Y.H., and C.S. wrote the manuscript. All authors agreed to submit the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yu Hu, Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1277 Jiefang Ave, Wuhan 430022, China; e-mail: dr_huyu@126.com; Chunyan Sun, Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1277 Jiefang Ave, Wuhan 430022, China; e-mail: suncy0618@163.com; Jian Li, Department of Hematology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1 Shuaifuyuan, Dongcheng District, Beijing, 100730, China; e-mail: lijian@pumch.cn; and Liqiong Liu,Department of Hematology, Huazhong University of Science and Technology Union Shenzhen Hospital, Shenzhen 518052, China; e-mail: llqwsp@hotmail.com.


