Key Points
How TPEx impacts rituximab effectiveness in iTTP patients is not fully understood.
In iTTP patients on therapeutic plasma exchange, rituximab eliminates circulating CD20+ B and T cells in 24 hours for at least 1 week.
Introduction
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a life-threatening thrombotic microangiopathy associated with severe deficiency in ADAMTS13, a disintegrin and metalloprotease responsible for limiting abnormally high concentrations of ultra-large von Willebrand factor multimers in the plasma of humans.1,2 Classically, iTTP results from B-cell production of immunoglobulin G (IgG) autoantibodies to ADAMTS13, leading to critically low activity levels (<10%) and formation of platelet-rich thrombi with thrombocytopenia, hemolytic anemia, and schistocytes under high-shear conditions in the microvasculature.3 Management of iTTP involves safely and quickly initiating immunomodulating agents targeting rogue lymphocytes responsible for IgG production alongside a treatment backbone of therapeutic plasma exchange (TPEx), which repletes ADAMTS13 and removes autoantibodies.4
Rituximab, a humanized mouse anti-CD20 monoclonal IgG1ĸ antibody, is commonly used in treatment regimens for iTTP and can prevent relapse and reduce mortality.5-8 Its half-life is ∼2 to 3 weeks but ultimately depends on the underlying treated condition, presence of TPEx, and CD20+ lymphocyte load. Rituximab eliminates CD20+ lymphocytes through multiple mechanisms, including antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and apoptosis.9 Data on rituximab pharmacokinetics during concurrent TPEx are limited, and the exact dosing, timing, and number of rituximab doses necessary to accomplish a durable, robust clinical response in the acute setting alongside ongoing TPEx, which can impact its clearance, is unknown and remains a topic of debate for iTTP.9-11 Although rituximab is believed to primarily target those CD20+ B cells inline to be the next generation of antibody-producing cells responsible for driving the autoimmune process, its effect on circulating CD20+ T cells in iTTP has not been reported. Importantly, although their function is debated, CD20+ T cells have been described in both healthy subjects and patients with other autoimmune conditions and are known to express cytokines and infiltrate human lymphatic tissues in patients with autoimmune diseases.12-16
Case description
In this prospective study, we describe our experience with administering rituximab during ongoing daily TPEx to 3 consecutive adult patients diagnosed with iTTP, each with a unique presentation.17 No significant side effects from rituximab administration were observed and we further present evidence that rituximab quickly and successfully eliminates circulating CD20+ B cells and a CD20+ subpopulation of T cells within 24 hours of rituximab dosing, which is sustained for at least 1 week with uninterrupted daily TPEx.
Methods
Three patients were prospectively enrolled. Whole blood was obtained in EDTA tubes under institutional research board–approved protocols in accordance with the Declaration of Helsinki. Blood samples were obtained at clinical diagnosis, after TPEx, and just prior to rituximab (day 0); 24 hours after dose 1 of rituximab and immediately post-TPEx (day 1); and just prior to rituximab dose 2 (day 7) (supplemental Figure 1). Platelet counts and other hematological parameters were followed systematically for each patient.
Peripheral blood mononuclear cells were enriched from 3 to 5 mL of EDTA-anticoagulated blood by red cell lysis. B-cell, T-cell, and natural killer cell compartments were analyzed using the following antibody combinations as previously described.18 Antibodies were used according to the manufacturers’ instructions and purchased from Becton Dickinson (San Jose, CA) unless otherwise noted. The antibody combinations were CD3–fluorescein isothiocyanate (FITC)/CD4-phycoerythrin (PE)/CD8-allophycocyanin (APC)/CD45–peridinin chlorophyll protein (PerCP), CD3-FITC/CD20-PE/CD19-APC/CD45-PerCP, and CD16-FITC (Beckman Coulter, Indianapolis, IN)/CD56-PE/CD3-APC/CD45-PerCP. Data were acquired using a FACSCanto cytometer and analyzed using BD FACSDiva version 8.0.1 (BD Biosciences, San Jose, CA). ADAMTS13 activity and inhibitor titers were determined at a referral laboratory (Versiti, Milwaukee, WI) using a modified fluorescence resonance energy transfer substrate–VWF73 assay and mixing studies, as previously described.19,20
Results and discussion
Patient characteristics are described in Table 1. Additional laboratory results and courses of patients are reported in supplemental Figures 2-5. Briefly, patient 1 (P1) had known recurrent iTTP, with multiple relapses in the past. Laboratory results and peripheral blood (PB) smear were consistent with relapsed iTTP. The patient sustained remission in 2 weeks with TPEx, steroids, and rituximab treatment.
Patient 2 (P2) was diagnosed with iTTP in the setting of metastatic melanoma treated with checkpoint immunotherapy ipilimumab/nivolumab. The last cycle was 41 days prior to admission. Laboratory results and PB smear were consistent with iTTP. After initially responding to therapy, by day 6, the patient had become refractory and obtained a second response after 22 days with prolonged TPEx, steroids, and rituximab. ADAMTS13 activity never normalized and the patient expired 154 days from diagnosis from complications of melanoma progression and iTTP. Patient 3 (P3) was diagnosed with iTTP in the setting of treatment with palliative temozolomide for metastatic ependymoma. Laboratory results and PB smear were consistent with iTTP. Although ADAMTS13 activity levels improved, initially platelets did not fully respond despite 4 weeks of a combination of TPEx, steroids, rituximab, and bortezomib.
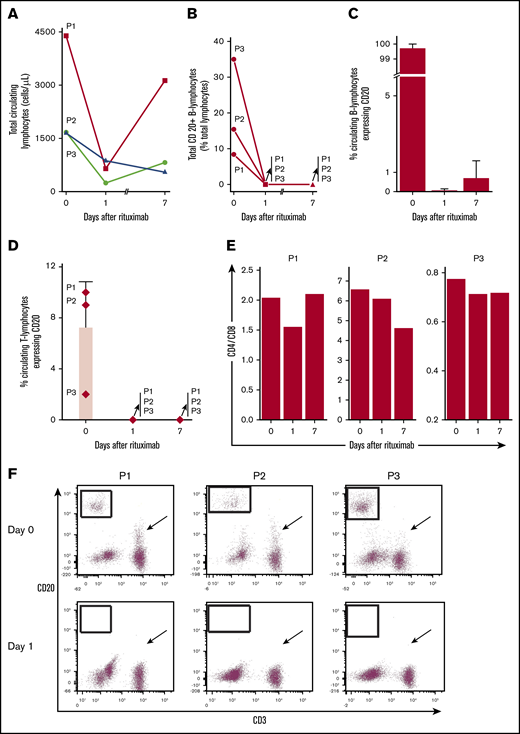
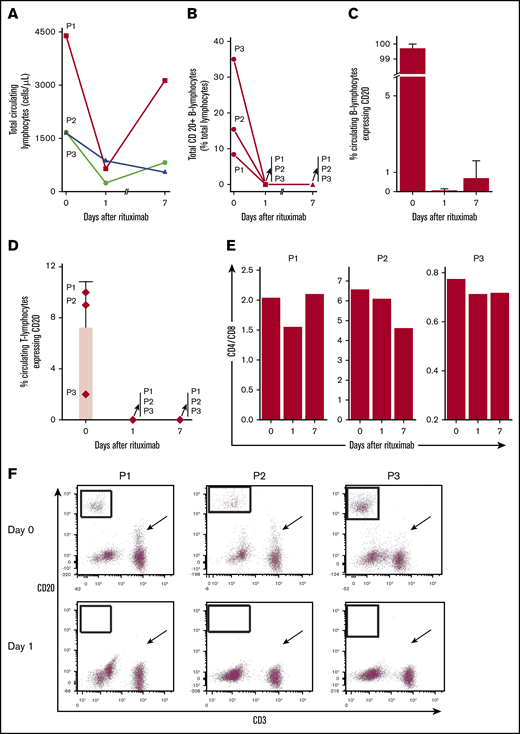
Total lymphocytes decreased after the first dose of rituximab (Figure 1A). CD20+ B lymphocytes represented 8% to 35% of circulating lymphocytes and most circulating B cells were CD20+ (Figure 1B-C). Rituximab eliminated essentially all circulating CD20+ B cells within 24 hours for each patient, despite uninterrupted daily TPEx. CD20+ B cells remained nearly absent 1 week later at the next rituximab dose. Rituximab also eliminated a CD20+ subpopulation of T cells in all 3 patients. The CD4-to-CD8 ratio concomitantly decreased from day 0 to day 1 (Figure 1D-F), suggesting preferential elimination of CD4+ T cells. The reduction in total T cells was greater than that which could be expected from eliminating just CD20+ T cells. Although CD20+ T cells accounted for a small proportion of all T cells, on average 71.4% (range, 38.5% to 88.3%) of all circulating T cells were eliminated among patients on day 1 (supplemental Figure 3). The CD3+20+ reduction persisted 1 week later as patients continued TPEx. The least responsive patient (P3) had the fewest CD20+ T cells, whereas the most responsive patient (P1) had the highest in circulation.
Rituximab leads to rapid and sustained elimination of circulating CD20+B and T lymphocytes despite ongoing TPEx in patients with iTTP. (A) Total circulating lymphocytes in patients with iTTP decrease after rituximab administration but there remains a variable response between the first and second dose of rituximab (days 1-7) and during continued TPEx. (B) Circulating CD20+ B lymphocytes vary in number in each patient. Rituximab removes nearly all circulating CD20+ B lymphocytes in each patient even after TPEx 24 hours later. The CD20+ B cells remain absent a week later at the time for the next rituximab treatment. (C) CD20+ is expressed on most circulating B lymphocytes in each patient at diagnosis. Rituximab quickly removes circulating CD20+ B lymphocytes within 24 hours. Recovery of CD20+ B lymphocytes by day 7 is patient-dependent, with patient 3 (P3) having the most circulating cells. (D) Specific circulating T lymphocytes in iTTP express CD20 and levels depend on the patient. Rituximab effectively depletes CD20+ T cells, which last 1 week after administration. (E) CD4-to-CD8 ratios are decreased between day 0 and day 1 in all patients, suggesting preferential depletion of CD4+ T cells. (F) Flow cytometry plots demonstrate that CD20+ B cells were eliminated (boxed regions) in each patient after rituximab treatment between day 0 and 1. The number of CD20+ T cells dropped dramatically (although to variable extent in different patients) within 24 hours after rituximab treatment (arrows). Bars and lines represent mean percentages plus standard deviation from N = 3 patients.
Rituximab leads to rapid and sustained elimination of circulating CD20+B and T lymphocytes despite ongoing TPEx in patients with iTTP. (A) Total circulating lymphocytes in patients with iTTP decrease after rituximab administration but there remains a variable response between the first and second dose of rituximab (days 1-7) and during continued TPEx. (B) Circulating CD20+ B lymphocytes vary in number in each patient. Rituximab removes nearly all circulating CD20+ B lymphocytes in each patient even after TPEx 24 hours later. The CD20+ B cells remain absent a week later at the time for the next rituximab treatment. (C) CD20+ is expressed on most circulating B lymphocytes in each patient at diagnosis. Rituximab quickly removes circulating CD20+ B lymphocytes within 24 hours. Recovery of CD20+ B lymphocytes by day 7 is patient-dependent, with patient 3 (P3) having the most circulating cells. (D) Specific circulating T lymphocytes in iTTP express CD20 and levels depend on the patient. Rituximab effectively depletes CD20+ T cells, which last 1 week after administration. (E) CD4-to-CD8 ratios are decreased between day 0 and day 1 in all patients, suggesting preferential depletion of CD4+ T cells. (F) Flow cytometry plots demonstrate that CD20+ B cells were eliminated (boxed regions) in each patient after rituximab treatment between day 0 and 1. The number of CD20+ T cells dropped dramatically (although to variable extent in different patients) within 24 hours after rituximab treatment (arrows). Bars and lines represent mean percentages plus standard deviation from N = 3 patients.
In this prospective study, we demonstrate that rituximab can be given safely to patients after TPEx and that it quickly and efficiently depletes CD20+ lymphocytes in patients with iTTP. It is the first study to demonstrate prospectively that CD20+ B cells are depleted from the circulation of patients with iTTP within 24 hours of rituximab despite ongoing daily TPEx, and CD20+ T cells are present in the PB of patients with iTTP and that these cells, too, are eliminated. Previously described as a polyclonal T-cell population often enriched in CD8+ and CD45RO+ memory T cells with a proinflammatory T helper type 1/cytotoxic T cell phenotype and high proliferative capacity to autoantigens, CD20+ T cells can also infiltrate primary and secondary lymphoid tissues and produce cytokines including interferon γ, tumor necrosis factor α, interleukin 4, and interleukin 17.12,13,16,21 Interestingly, our results suggest that CD4+ T cells too may play a role in rituximab response during iTTP treatment. With the reduction in PB CD4-to-CD8 ratio post–rituximab treatment, some of the lost T cells may be follicular helper CD4+ T cells, which depend on B cells and play a role in autoimmune diseases.22 Findings of CD20+ T cells in circulation of patients with iTTP, along with previous descriptions of CD20+ T cells present in other autoimmune conditions,16 also support that these T cells may contribute to the pathogenesis of iTTP. Furthermore, recent evidence suggests that lymphocytes with tandem T- and B-cell markers can disrupt the paradigm of absolute compartmentalization of adaptive immunity and may act as drivers of autoimmunity.23 CD20+ T lymphocytes may also uphold these findings. Furthermore, our report reinforces that immunotherapies using checkpoint blockade to treat cancer can induce life-threatening autoimmune hematological conditions, including iTTP.24,25 Additional investigation is needed to elucidate the role CD20+ T cells play in iTTP pathophysiology and whether our findings can be replicated with changes in dosing and/or frequency of administration of rituximab.
For original data, please contact bodoimre.md@gmail.com.
Acknowledgments
This work was supported by the National Institutes of Health through National Center for Advancing Translational Sciences grant KL2TR002490 (M.A.C.) and the Hungarian National Research Development and Innovation Office (NFKI) grant OTKA-K19_131945 (I.B.).
Authorship
Contribution: M.A.C. collected clinical data and patient samples, interpreted the data, performed statistical analyses, and wrote the manuscript; I.B. and D.L.J. designed the study, collected samples, interpreted data, and cowrote the manuscript; M.G. and R.K. collected clinical data and carefully read the manuscript; A.H., M.B., V.M., and S.R.S. assisted in collecting clinical samples, provided patient care, and carefully read and edited the manuscript; A.A. and S.C. assisted in the literature search, provided expert opinion, and carefully read and edited the manuscript; and D.L.J. and S.K. performed flow cytometric analyses.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Imre Bodó, 3rd Department of Medicine, Semmelweis University, Kútvölgyi út 4, 1125 Budapest, Hungary; e-mail: bodoimre.md@gmail.com.