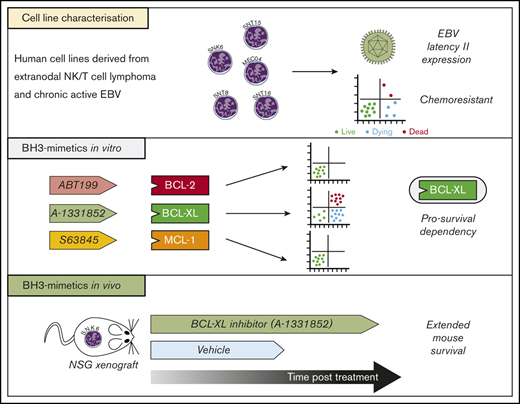
Key Points
Representative ENKTL and CAEBV human cell lines, with diverse TP53 status and MDR1 activity, are insensitive to chemotherapeutic drugs.
Blockade of BCL-XL alone induces killing of ENKTL and CAEBV cells but is most effective when MCL-1 is additionally targeted.

Abstract
Epstein-Barr virus (EBV)–associated T- and natural killer (NK)–cell malignancies, such as extranodal NK-/T-cell lymphoma (ENKTL), exhibit high chemoresistance and, accordingly, such patients have a poor prognosis. The rare nature of such cancers and nonmalignant T/NK lymphoproliferative disorders, such as chronic active EBV (CAEBV), has limited our understanding of the pathogenesis of these diseases. Here, we characterize a panel of ENKTL- and CAEBV-derived cell lines that had been established from human tumors to be used as preclinical models of these diseases. These cell lines were interleukin-2 dependent and found to carry EBV in a latency II gene-expression pattern. All cell lines demonstrated resistance to cell death induction by DNA damage–inducing agents, the current standard of care for patients with these malignancies. This resistance was not correlated with the function of the multidrug efflux pump, P-glycoprotein. However, apoptotic cell death could be consistently induced following treatment with A-1331852, a BH3-mimetic drug that specifically inhibits the prosurvival protein BCL-XL. A-1331852–induced apoptosis was most efficacious when prosurvival MCL-1 was additionally targeted, either by BH3-mimetics or genetic deletion. Xenograft models established from the ENKTL cell line SNK6 provided evidence that A-1331852 treatment could be therapeutically beneficial in vivo. The data here suggest that therapeutic targeting of BCL-XL would be effective for patients with EBV-driven T/NK proliferative diseases, however, MCL-1 could be a potential resistance factor.
Introduction
Extranodal natural killer (NK)–/T-cell lymphoma (ENKTL) is an aggressive malignancy predominantly presenting within the upper aerodigestive tract as a highly necrotic tumor with invasion into local tissue.1 ENKTL is a relatively rare disease but found with a higher frequency in eastern Asia and South America (4% to 6% of non-Hodgkin lymphomas) compared with western populations (0.5% of non-Hodgkin lymphomas).2 Chemotherapy regimens for ENKTL, such as cyclophosphamide, hydroxydaunorubicin, vincristine, prednisone (CHOP), are largely ineffective, yielding a median patient survival of only 7.8 months.3 Such poor effectiveness of CHOP in ENKTL has been attributed to expression and function of the multidrug efflux pump, P-glycoprotein/MDR1.4,5 Accordingly, a new chemotherapy regimen with theoretically reduced MDR1 susceptibility (dexamethasone, methotrexate, ifosfamide, l-asparaginase, etoposide [called SMILE]) has been developed and has shown promise in early small cohort studies.6-8 Of note, severe and prolonged cases of an Epstein-Barr virus (EBV)–driven nonmalignant T/NK-cell proliferative disease, called chronic active EBV (CAEBV), can occasionally progress into ENKTL.9 Here, chemotherapy can only be effective when combined with hematopoietic stem cell transplantation during early stages of disease.10,11
ENKTLs are typically NK cell in origin, although a minority of cases are of T-cell lineage, with a characteristic immunophenotype of CD2+, CD56+, and cytoplasmic CD3ε+ but surface CD3−.12-14 Disease etiology is poorly understood but deletions at chromosome 6p21-q25 are the most frequent chromosomal abnormality in ENKTL (40% to 50% of cases based on small patient studies15,16 ). The main target of this deletion is thought to be the transcriptional repressor PRDM1 but consistent genetic drivers of disease are yet to be identified.15 In CAEBV, however, a study of 80 patient samples identified mutations in the tumor repressor DDX3X as the most common disease driver with a mutation frequency of 17.5%.17 A confounding factor is that, in 100% of ENKTL cases, the malignant cells are infected with EBV, a principally B-lymphotropic herpesvirus. This requisite association indicates a critical role for EBV as a driver of this disease, although a mechanism of T/NK-cell transformation by EBV is not yet clearly described.1
EBV is ubiquitous within the human population where the virus resides lifelong in memory B cells, typically as an asymptomatic infection. Despite its widespread prevalence, EBV is an oncogenic virus responsible for ∼200 000 new malignancies annually, usually of B-cell, epithelial cell, or T/NK-cell origin.18 In malignancy, EBV displays a predominantly latent form of infection, involving all, or a subset, of latent genes/transcripts termed EBV nuclear antigens (EBNA1, 2, 3A, 3B, 3C, and LP), the latent membrane proteins (LMP1, 2A, and 2B), noncoding EBV-encoded small RNA (EBER) transcripts and several microRNAs.19 These viral products have been extensively studied in B-cell and epithelial cell backgrounds with respect to cellular transformation, induction of proliferation, and maintenance of cell survival,20 but are less well studied in T/NK cells. Within ENKTL and CAEBV cells, EBV typically expresses a latency II restricted gene-expression pattern: EBNA1+, LMP1+, and/or LMP2A/B+, in addition to noncoding viral RNAs.19,21-23 Importantly, the major viral oncogene, LMP1, is reportedly expressed in 73% of ENKTL tumors.24 A key component of LMP1 activity in B and epithelial cells is that it potently stimulates the NF-κB pathway, which can lead to upregulation of BCL-2 family prosurvival proteins BCL-2, MCL-1, and BFL-1.25-27
The intrinsic apoptotic pathway is regulated by the BCL-2 family where the balance of specific interactions between pro- and antiapoptotic members decides the fate of a cell. The antiapoptotic proteins (BCL-2, BCL-XL, BCL-W, MCL-1, and A1/BFL1) prevent the proapoptotic effector proteins (BAK and BAX) from inducing mitochondrial outer membrane permeabilzation (MOMP), the point of no return in apoptosis.28 Upon an apoptotic stimulus, proapoptotic BH3-only proteins (BIM, PUMA, NOXA, BID, BMF, BIK, BAD, and HRK) are increased (through transcriptional or posttranscriptional processes), bind with very high affinity to the antiapoptotic members, and thereby unleash the proapoptotic effectors BAX and BAK to induce MOMP.29 Select BH3-only proteins can also directly bind and thereby activate BAX and BAK.29
Deregulation of the intrinsic apoptotic pathway is common in tumor cells and confers protection from cell death induced by oncogenic stress or chemotherapy.30 Such deregulation can occur through increased expression of antiapoptotic BCL-2 proteins (eg, Bcl-2/IgH chromosomal translocation in follicular B-cell lymphoma) or reduced expression of proapoptotic members (eg, Bim gene hypermethylation in Burkitt lymphoma).31-33 This, together with the knowledge that viral proteins can modulate expression of key cellular BCL-2 proteins,20 led us to speculate that targeting of select BCL-2 prosurvival proteins may offer a novel therapeutic option for ENKTL patients. Small-molecule inhibitors of BCL-2 prosurvival proteins, known as BH3-mimetics, have been developed to induce apoptosis in malignant cells.34 The BCL-2–specific inhibitor, ABT-199 (venetoclax), is approved by the US Food and Drug Administration (FDA) for treatment of chemotherapy-refractory chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML).35,36 Clinical trials have commenced with 3 MCL-1 targeting BH3-mimetic drugs in hematological cancers.37-40 BCL-XL–specific compounds, such as A-1331852, are less advanced but have shown success in preclinical models.41,42 Such studies, and data from clinical trials with ABT-263/navitoclax, which targets BCL-XL, BCL-2, and BCL-W, indicate a narrow therapeutic window for BCL-XL inhibitors due to dose-limiting on-target thrombocytopaenia.43
Here, we characterize a panel of ENKTL and CAEBV patient-derived cell lines as appropriate preclinical disease models. Through in vitro analyses and an in vivo mouse xenograft model, we demonstrate that therapeutic targeting of BCL-XL alone, or in combination with MCL-1 inhibition, can have therapeutic efficacy for EBV-driven T- and NK-cell disease.
Materials and methods
Animal work
Experiments performed with mice were conducted in accordance with guidelines of The Walter and Eliza Hall Institute Animal Ethics Committee. Single-cell suspensions of ENKTL/CAEBV cell lines were delivered to 6- to 8-week-old NOD-SCID-IL2rg−/− (NSG) female mice44 by subcutaneous (2 × 106 cells) or intraperitoneal (5 × 106 cells) injection.
A-1331852, formulated in 60% Phosal 50 PG (Lipoid), 27.5% polyethylene glycol 400 (Sigma), 10% ethanol, and 2.5% dimethyl sulfoxide (DMSO) was administered by oral gavage at 50 mg/kg once daily for 7 consecutive days. Upon fulfillment of end point criteria, mice were euthanized by CO2 exposure.
Cell lines and cytogenetic analyses
Cell lines SNK6, SNT8, SNT15, SNT16, and MEC0445-48 were grown in RPMI 1640 medium (Sigma) supplemented with 10% vol/vol heat-inactivated and sterile-filtered human serum (Sigma), 100 U/mL penicillin (Invitrogen), 100 µg/mL streptomycin (Invitrogen), 6 mM l-glutamine (Invitrogen), 1 mM sodium pyruvate (Sigma), 700 U/mL IL-2 (Novartis). BL-41 and Raji cells were cultured in RPMI 1640 medium supplemented with 10% vol/vol FCS, 1 mM glutamine (Invitrogen), 1 mM sodium pyruvate (Sigma), and 50 µM thioglycerol (Sigma). All T/NK-cell lines were karyotyped as described in supplemental Methods.
Targeted TP53 gene sequencing
DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions. Amplicon libraries targeting TP53 exons 2-11 (NM_000546) were prepared using a 2-step polymerase chain reaction (PCR) approach: first internal PCR to amplify the regions of interest (supplemental Table 1), followed by second outer PCR to add the sequencing adaptors, and indexes for multiplexing as detailed in supplemental Methods.
Western blotting
Total protein extracts were prepared from cells by lysis using radioimmunoprecipitation assay (RIPA) buffer containing complete mini protease inhibitor cocktail (Roche). Western blotting was performed as detailed in supplemental Methods.
Cell-death assays
Cells were exposed to drugs (supplemental Table 3) at the specified doses, harvested at set time points, and stained with annexin V (conjugated to fluorescein isothiocyanate [FITC] or Alexa Fluor 647) and propidium iodide (2 µg/mL) in annexin V binding buffer (0.1 M N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid [HEPES; pH 7.4], 1.4 M NaCl, 25 mM CaCl2). Samples were analyzed in an LSR-II, LSR-Fortessa X-20, or Accuri C6 (BD Biosciences) flow cytometer and data analyzed using FlowJo 10 (Treestar).
Cell-surface phenotyping
Cells were first stained with viability dye (violet stain) (ThermoFisher) according to the manufacturer’s instructions and subsequently stained for surface markers by incubating on ice for 30 minutes with saturating concentrations of antibodies described in supplemental Table 4. Cells were analyzed on a LSR Fortessa II (BD Biosciences) and data were processed using FlowJo software (Treestar).
Rhodamine-123 efflux assay
Expression and functionality of P-glycoprotein was determined by measuring the relative intensity of cell staining using the fluorescent molecule rhodamine-123 (Rh-123; Sigma), a P-glycoprotein–specific substrate, as described in the supplemental Methods.
RNA extraction and cDNA synthesis
RNA was prepared using Nucleospin II kit (Macherey-Nagel) and DNAse I treated using a DNA-free kit (Life Technologies) and complementary DNA (cDNA) generated using QScript (VWR), all according to the manufacturer’s protocols.
Quantitative reverse transcriptase (qRT)–PCR analysis for EBV transcripts
Absolute quantification of EBV gene transcripts by high throughput quantitative PCR was performed using the Fluidigm Dynamic Array system as previously described49 and detailed in supplemental Methods.
CRISPR/Cas9 gene editing
CRISPR/Cas9-induced knockout of MCL-1 in MEC04 cells was induced using an inducible lentiviral guide RNA system as previously described.50 Sequences of single-guide RNAs (sgRNAs) are as follows: human MCL-1 exon 1, 5′-GGGAGGGCGACTTTTGGCTA; mouse Bim exon 2 (control sgRNA), 5′-GAGCGCTGCTCCGATGGTGA.
Statistical analysis
Prism software (GraphPad) was used to perform statistical tests with P < .05 considered significant. Data presented are means of 3 independent experiments unless stated otherwise. Error bars indicate the standard deviation or standard error as indicated in figure legends.
Results
Characterizing EBV+ T- and NK-cell lines
Although reports assessing patient samples have been published, it should be noted that general availability of primary material is extremely limited.51,52 Due to the rarity of EBV-driven T- and NK-cell lymphoproliferative diseases and the difficulties to derive immortalized cell lines, attributed to the necrotic nature of ENKTL tumors, most functional studies described within the literature have focused on only a subset of the few available ENKTL and CAEBV cell lines. Here, we characterize a comprehensive collection of 3 ENKTL cells lines (SNK6, SNT8, and MEC04) and 2 CAEBV cell lines (SNT15 and SNT16), to determine how closely they represent these diseases.45-48
Consistent with their reported NK-cell phenotype, all cells within SNK6 and MEC04 cell populations expressed CD56 (supplemental Figure 1). As expected, SNT8 and SNT15 cells were positive for TCR Vd2, however, SNT15 cells were CD56−. The CAEBV cell line SNT16, was of CD4 T-cell lineage, as reported.47 All cell lines require interleukin 2 (IL-2) for continued proliferation in vitro and, accordingly, they all expressed the IL-2 receptor α chain (CD25), although MEC04 cells expressed comparatively low levels.
EBV gene expression was quantified by absolute qRT-PCR assays. Transcripts specific for latent EBNA1 and LMP2 were detected in all cell lines (Figure 1A). Transcripts and LMP1 protein (revealed by western blotting) could only be detected in SNK6, SNT8, and SNT16 cells (Figure 1B). The absence of EBNA2 transcripts and protein in all cell lines indicated a latency II viral gene-expression pattern (Figure 1B). Lytic cycle-associated transcripts could be detected at low levels in all cell lines, with the highest level of BZLF1, a lytic cycle transcriptional activator, detected in SNT16 cells at 0.0665 transcripts per phosphoglycerate kinase (PGK; Figure 1A). In a 100% lytic cellular population, the expected level of BZLF1 exceeds 9 copies per PGK,49 suggesting the levels observed in the T/NK-cell lines are a consequence of spontaneous abortive EBV lytic activation in rare cells, a common phenomenon in EBV+ cell lines.19 The apparent latency II gene-expression pattern and lack of lytic activity observed within the T/NK-cell line panel is comparable to patient CAEBV and ENKTL samples.53

EBV displays a latency II gene-expression pattern in ENTKL- and CAEBV-derived cell lines. (A) Heatmap summary of EBV transcript levels to compare between the different cell lines. Data shown were normalized to the levels of PGK1 transcripts. (B) Expression levels of key viral proteins, LMP1 and EBNA2, at steady state were assessed by western blot analysis. Probing for HSP70 was used as a protein loading control and BL cell lines, BL41 and Raji, and a lymphoblastoid cell line (LCL) were included as controls. Representative blots are shown.
EBV displays a latency II gene-expression pattern in ENTKL- and CAEBV-derived cell lines. (A) Heatmap summary of EBV transcript levels to compare between the different cell lines. Data shown were normalized to the levels of PGK1 transcripts. (B) Expression levels of key viral proteins, LMP1 and EBNA2, at steady state were assessed by western blot analysis. Probing for HSP70 was used as a protein loading control and BL cell lines, BL41 and Raji, and a lymphoblastoid cell line (LCL) were included as controls. Representative blots are shown.
Resistance to cytotoxic agents is not universally mediated by MDR1 activity or TP53 mutations
Considering the apparent lack of response to standard-of-care chemotherapeutic drugs in ENKTL patients, we may expect that ENKTL and perhaps CAEBV-derived cell lines will display resistance to such compounds in vitro. Following a 48 hours treatment with etoposide, 2 cell lines (SNK6 and SNT16) exhibited high resistance to apoptosis (50% inhibitory concentration [IC50] values > 10 μM) and the remaining cell lines (SNT8, SNT15, and MEC04) exhibited moderate resistance (IC50 values of 1.9 μM, 3.3 μM, and 7.2 μM, respectively) (Figure 2A). Similarly, methotrexate treatment had only modest impact, with 4 of 5 cell lines displaying high levels of resistance (IC50 values > 10 μM) (Figure 2B). In fact, even for MEC04 cells, that had comparable IC50 values to the susceptible control cell line MOLT4 (1.2 μM and 1.1 μM, respectively), there remained 42.3% viability at the highest tested concentration (10 μM), compared with only 6.1% for MOLT4 cells. This indicates that a significant proportion of the MEC04 cell population were highly resistant to methotrexate. These drugs act through TP53 but sequencing revealed wild-type TP53 sequences in all cell lines, with the exception of MEC04 that had a c.743G>A (p.Arg248Gln) point mutation in exon 7 (Table 1).

Resistance to common chemotherapeutic drugs in ENKTL and CAEBV cell lines is not only due to P-glycoprotein–mediated drug efflux. Dose-response curves generated from annexin V/propidium iodide (PI) staining assays following 48-hour treatment with etoposide (A) or methotrexate (B) at the indicated concentrations. (C) The functionality of P-gp was assessed by the efflux of fluorescent rh-123 using flow cytometry. Cells were either left unstained or stained with rh-123 (black, filled histogram), stained with subsequent incubation at 4°C (gray, filled histogram), stained with subsequent incubation at 37°C (red, open histogram) or stained with subsequent incubation at 37°C in the presence of the P-gp inhibitor, CsA (blue, open histogram). (D) Rh-123 efflux was quantified using the following formula: (MFIred × 100) / (MFIgray − MFIblack). KG-1 cells (positive control) and Jurkat cells (negative control) were included in the experiments due to their reported high P-gp expression and lack of P-gp expression, respectively. Data are presented as means ± standard deviation (SD) of 3 independent experiments, each performed in triplicate (n = 3). ***P < .0001, vs Jurkat. CsA, cyclosporin A; MFI, median fluorescence intensity; P-gp, P-glycoprotein.
Resistance to common chemotherapeutic drugs in ENKTL and CAEBV cell lines is not only due to P-glycoprotein–mediated drug efflux. Dose-response curves generated from annexin V/propidium iodide (PI) staining assays following 48-hour treatment with etoposide (A) or methotrexate (B) at the indicated concentrations. (C) The functionality of P-gp was assessed by the efflux of fluorescent rh-123 using flow cytometry. Cells were either left unstained or stained with rh-123 (black, filled histogram), stained with subsequent incubation at 4°C (gray, filled histogram), stained with subsequent incubation at 37°C (red, open histogram) or stained with subsequent incubation at 37°C in the presence of the P-gp inhibitor, CsA (blue, open histogram). (D) Rh-123 efflux was quantified using the following formula: (MFIred × 100) / (MFIgray − MFIblack). KG-1 cells (positive control) and Jurkat cells (negative control) were included in the experiments due to their reported high P-gp expression and lack of P-gp expression, respectively. Data are presented as means ± standard deviation (SD) of 3 independent experiments, each performed in triplicate (n = 3). ***P < .0001, vs Jurkat. CsA, cyclosporin A; MFI, median fluorescence intensity; P-gp, P-glycoprotein.
MDR1 expression has been proposed to be responsible for chemotherapy resistance in ENKTL tumors.4,5 To examine this, MDR1 activity was assessed by efflux of the MDR1-specific substrate rhodamine-123 in the ENKTL and CAEBV cell lines. Rhodamine-123 efflux was observed in the NK-ENKTL cell lines SNK6 and MEC04, but absent in the T-ENKTL cell line, SNT8, and in both CAEBV cell lines (Figure 2C). Collectively, these data indicate that neither MDR1 activity nor TP53 mutations were consistent mediators of the observed resistance of EBV+ T- and NK-cell lines to genotoxic anticancer agents.
BCL-XL–specific BH3-mimetic drugs consistently induced apoptosis in ENKTL and CAEBV cell lines
Downstream of TP53, the intrinsic apoptotic pathway must be functional for induction of cell death by genotoxic agents. Expression of prosurvival proteins BCL-2, BCL-XL, and MCL-1 were universally detected, where BCL-2 showed the most consistent expression in the panel of ENKTL and CAEBV cell lines examined (Figure 3A). Proapoptotic BIM and BID expression was absent or low in most cell lines relative to the control cell line BL41. The proapoptotic effectors, BAK and BAX, were universally expressed. Taken together, the expression data indicate that theoretically all cell lines should be capable of undergoing intrinsic apoptosis.

BH3-mimetic drugs targeting BCL-XL induce cell death in ENKTL and CAEBV cell lines. (A) Expression levels of cellular BCL-2, BCL-XL, MCL-1, BIM, PUMA, NOXA, BAK, and BAX were detected by western blot analysis. Probing for HSP70 was used as a protein loading control. The Burkitt lymphoma–derived cell lines, BL41 and Raji, and LCL were used as controls. Representative blots are shown. (B) Cells were treated for 48 hours with 0.5 µM or 5 µM of the indicated BH3-mimetic drugs ABT-199 (BCL-2 specific), ABT-737 (inhibits BCL-2, BCL-XL, and BCL-W), A-1331852 (BCL-XL specific), or S63845 (MCL-1 specific). Cells were then stained with annexin V and PI and their viability quantitated by flow cytometry. Data shown are normalized to DMSO-treated control cells and presented as mean ± SD of 3 independent experiments.
BH3-mimetic drugs targeting BCL-XL induce cell death in ENKTL and CAEBV cell lines. (A) Expression levels of cellular BCL-2, BCL-XL, MCL-1, BIM, PUMA, NOXA, BAK, and BAX were detected by western blot analysis. Probing for HSP70 was used as a protein loading control. The Burkitt lymphoma–derived cell lines, BL41 and Raji, and LCL were used as controls. Representative blots are shown. (B) Cells were treated for 48 hours with 0.5 µM or 5 µM of the indicated BH3-mimetic drugs ABT-199 (BCL-2 specific), ABT-737 (inhibits BCL-2, BCL-XL, and BCL-W), A-1331852 (BCL-XL specific), or S63845 (MCL-1 specific). Cells were then stained with annexin V and PI and their viability quantitated by flow cytometry. Data shown are normalized to DMSO-treated control cells and presented as mean ± SD of 3 independent experiments.
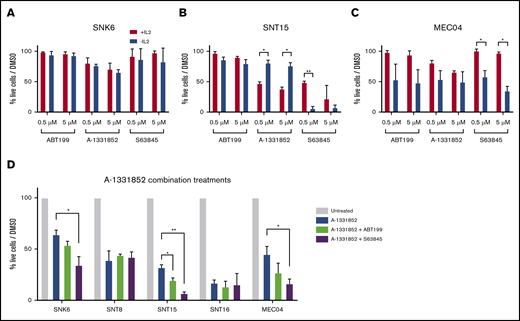
The sustained growth and survival of several cancers has been attributed to reliance on 1 or more of the prosurvival proteins, BCL-2, BCL-XL, and MCL-1.36,54-58 To investigate this in the context of ENKTL and CAEBV, cell lines were treated with various BH3-mimetic drugs (Figure 3B). Despite universal expression of BCL-2, the cell lines were resistant to treatment with the BCL-2–specific inhibitor ABT-199. They displayed similar resistance to the MCL-1 inhibitor S63845, with the exception of the SNT15 cells. The greatest sensitivity observed across all cell lines was for treatment with the BCL-XL–specific compound, A-1331852. Accordingly, ABT-737, which targets BCL-XL, BCL-2, and BCL-W, also induced apoptosis across the cell line panel, consistent with the EBV+ T/NK-cell lines being BCL-XL dependent. Interestingly, the sensitivity to BCL-XL inhibition was independent of whether the cell lines expressed viral LMP1.
The BCL-XL inhibitor A-1331852 delays tumor growth in ENKTL-engrafted NSG mice
As A-1331852 has been demonstrated to be bioavailable in animal models of breast, ovarian and small cell lung cancer,41 we examined its impact on tumor growth in in vivo models of ENKTL and CAEBV. SNK6, SNT15, and MEC04 cells were either subcutaneously or intraperitoneally injected into NOD-SCID-IL2rg−/− (NSG) recipient mice followed by daily treatments with 50 mg/kg A-1331852 for 7 days. In NSG mice intraperitoneally injected with SNK6 cells (SNK6-IP mice), a significant delay in tumor development was observed after treatment with A-1331852 compared with vehicle-treated mice, with median tumor latencies of 56 days and 37 days posttransplant, respectively (P = .0002) (Figure 4A). Similarly, in SNK6-SC–bearing mice, A-1331852 treatment significantly delayed tumor growth (median tumor latency of 56 days compared with vehicle-treated mice (37 days) (P = .022)) (Figure 4B). In contrast, A-1331852 was ineffective at delaying tumor growth in mice transplanted with SNT15 cells (Figure 4C) or MEC04 cells (Figure 4D). Following drug treatment (day 14), there was a marked decrease in platelet numbers, consistent with successful administration of the drug with BCL-XL inhibition causing on-target thrombocytopenia (Figure 4A,D).59 As expected, the platelet counts rebounded to pretreatment levels by the time of sacrifice (days 34-62 posttransplant). Unfortunately, we were unable to assess the efficacy of BCL-XL inhibition in tumor growth using SNT8 and SNT16 cells as they did not reliably grow as xenografts in NSG mice.
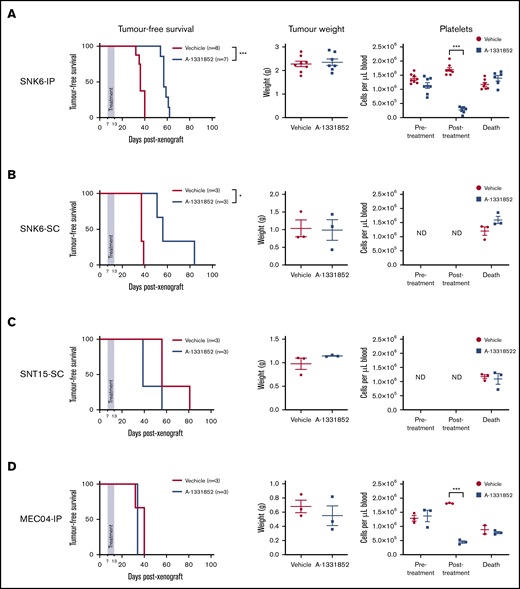
The BCL-XL–specific BH3-mimetic drug A-1331852 inhibits tumor growth in NSG mice that had been transplanted with SNK6 cells. NSG mice were injected subcutaneously (SC) or intraperitoneally (IP) with SNK6, SNT15, or MEC04 cells on day 0. From day 7, A-1331852 or vehicle were delivered once daily to mice by oral gavage for 7 consecutive days. Mice were then monitored for tumor burden. Platelet levels in peripheral blood were examined pretreatment (day 7), posttreatment (day 14), and immediately prior to euthanization. NSG mice were injected intraperitoneally with SNK6 cells (5 × 106 cells) (A), subcutaneously with SNK6 cells (2 × 106 cells) (B), subcutaneously with SNT15 cells (2 × 106 cells) (C), or intraperitoneally with MEC04 cells (5 × 106 cells) (D). Mouse survival curves were assessed for significant differences by the Mantel-Cox test. Platelet and tumor weight data are shown with mean ± standard error (SE), and statistical significance were determined by unpaired Student t test. *P < .05; ***P < .0001. ND, not done.
The BCL-XL–specific BH3-mimetic drug A-1331852 inhibits tumor growth in NSG mice that had been transplanted with SNK6 cells. NSG mice were injected subcutaneously (SC) or intraperitoneally (IP) with SNK6, SNT15, or MEC04 cells on day 0. From day 7, A-1331852 or vehicle were delivered once daily to mice by oral gavage for 7 consecutive days. Mice were then monitored for tumor burden. Platelet levels in peripheral blood were examined pretreatment (day 7), posttreatment (day 14), and immediately prior to euthanization. NSG mice were injected intraperitoneally with SNK6 cells (5 × 106 cells) (A), subcutaneously with SNK6 cells (2 × 106 cells) (B), subcutaneously with SNT15 cells (2 × 106 cells) (C), or intraperitoneally with MEC04 cells (5 × 106 cells) (D). Mouse survival curves were assessed for significant differences by the Mantel-Cox test. Platelet and tumor weight data are shown with mean ± standard error (SE), and statistical significance were determined by unpaired Student t test. *P < .05; ***P < .0001. ND, not done.
IL-2 deprivation results in a reduction in BCL-XL levels and an increase in MCL-1 expression
Following xenotransplantation of SNT15 and MEC04 cells (Figure 4C-D), no delay in tumor growth was observed with A-1331852 treatment despite the marked sensitivity of these cells to this compound in vitro (Figure 3B). In vitro all ENKTL and CAEBV cell lines require exogenous IL-2 for continued survival and growth (Figure 5A). We speculated that the restricted IL-2 availability in NSG mice, due to the absence of IL-2–producing lymphocytes in this model, may have impacted the expression of BCL-2 family proteins. Therefore, to mimic the in vivo setting, we deprived cells of IL-2 for 48 hours in vitro, then assessed BCL-2 family protein expression. BIM expression increased in SNT15 and MEC04 (the only BIM-expressing cell lines) indicating that cells were responding to cytokine deprivation. Importantly, BCL-XL expression decreased in the absence of IL-2 (Figure 5B), whereas MCL-1 expression increased in all cell lines (Figure 5B; supplemental Figure 2).
![IL-2 withdrawal leads to loss of viability and reduced BCL-XL expression in ENKTL cell lines. (A) ENKTL cells cultured in medium lacking IL-2 were assessed over a 120-hour time course by flow cytometry for viability as determined by annexin V plus PI staining. Data were normalized to cell viability of cells grown in medium supplemented with IL-2 and are presented as means ± SD of 3 independent experiments, each performed in triplicates (n = 3). (B) Expression levels of BCL-XL, MCL-1, and BIM proteins were detected by western blot analysis in cells cultured in medium lacking IL-2 (in the presence of the broad-spectrum caspase inhibitor QVD-OPH [25 µM] to prevent late stages of apoptosis with cell demolition) at the indicated time points. Probing for HSP70 was used as a protein loading control. Representative blots are shown.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/19/10.1182_bloodadvances.2020002446/2/m_advancesadv2020002446f5.png?Expires=1763932387&Signature=45f1esTsqvXlm9KKAjnXjkaKdJGv7whMlRfJBxS82~4Jxi24JYnAQtihMLcdnWoV0bpVAjRiPgdNU-Cgzkq-NpLunU3ruWokaDez8Ipzve9BG9lh11SmSkGY~XuOfRUc6hrD5g~3WIBZvdDwAd0avvLrPIIWzprXF447WG0Yn7mxhpu6-mOYLUvz-NjL8UPaeftB2KFEXCr2N9Q2ewo2vSiMbEZa6aXy-tDoQnhkUPg59AYBbRTCYt3cWMaBqUy8lGVwz8e1ufCG8Jey4Qo2snNd8KBc-vTJ0BtQbAPR4XEk3mPCxND0rNYf6yEU-i9Cswdm0UiTWe4r73cAohCo0g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IL-2 withdrawal leads to loss of viability and reduced BCL-XL expression in ENKTL cell lines. (A) ENKTL cells cultured in medium lacking IL-2 were assessed over a 120-hour time course by flow cytometry for viability as determined by annexin V plus PI staining. Data were normalized to cell viability of cells grown in medium supplemented with IL-2 and are presented as means ± SD of 3 independent experiments, each performed in triplicates (n = 3). (B) Expression levels of BCL-XL, MCL-1, and BIM proteins were detected by western blot analysis in cells cultured in medium lacking IL-2 (in the presence of the broad-spectrum caspase inhibitor QVD-OPH [25 µM] to prevent late stages of apoptosis with cell demolition) at the indicated time points. Probing for HSP70 was used as a protein loading control. Representative blots are shown.
IL-2 withdrawal leads to loss of viability and reduced BCL-XL expression in ENKTL cell lines. (A) ENKTL cells cultured in medium lacking IL-2 were assessed over a 120-hour time course by flow cytometry for viability as determined by annexin V plus PI staining. Data were normalized to cell viability of cells grown in medium supplemented with IL-2 and are presented as means ± SD of 3 independent experiments, each performed in triplicates (n = 3). (B) Expression levels of BCL-XL, MCL-1, and BIM proteins were detected by western blot analysis in cells cultured in medium lacking IL-2 (in the presence of the broad-spectrum caspase inhibitor QVD-OPH [25 µM] to prevent late stages of apoptosis with cell demolition) at the indicated time points. Probing for HSP70 was used as a protein loading control. Representative blots are shown.
MCL-1 compensates for inhibition or downregulation of BCL-XL
BCL-XL was identified as the primary survival factor in ENKTL and CAEBV cells and its expression was changed by IL-2 withdrawal. We hypothesized that at least for SNT15 and MEC04 cells that could readily expand in NSG mice following A-1331852 treatment, and possibly for other cell lines also, a secondary factor must also contribute to survival in circumstances where BCL-XL is low/inhibited. Furthermore, IL-2 withdrawal led to a slow, rather than immediate, death of cells over many days after the initial decrease in BCL-XL expression and for some cell lines the viability then stabilized. To identify the secondary survival factor(s), BH3-mimetic drug treatment of SNK6, SNT15, and MEC04 cells was performed after 48 hours of culture in medium lacking IL-2 leading to reduced levels of BCL-XL. As in vivo, SNK6 cells maintained comparable sensitivity to A-1331852 in vitro when deprived of IL-2 (Figure 6A). In contrast, SNT15 cells displayed decreased sensitivity to A-1331852 in medium lacking IL-2 (Figure 6B), an observation in agreement with their in vivo resistance to BCL-XL inhibitor treatment (Figure 4D). MEC04 cells showed a trend toward increased sensitivity to A-1331852 following IL2 withdrawal, however this was not statistically significant (Figure 6C). Notably, a significant increase in sensitivity to the MCL-1 inhibitor S63845 was observed in both SNT15 and MEC04 cells when cultured without IL-2. This suggests that these cells display a dependency on MCL-1 under such stress conditions. Consistently, the combined in vitro treatment of A-1331852 with S63845 proved the most potent inducer of apoptosis in SNK6, SNT15 and MEC04 cells relative to other combinations of BH3-mimetics (Figure 6D). This further suggests that MCL-1 plays a critical role alongside BCL-XL in sustaining the survival of ENKTL and CAEBV cells.
Sensitivity of ENKTL cell lines to the MCL-1 inhibitor S63845 increases upon IL-2 withdrawal or treatment with the BCL-XL inhibitor A-1331852. SNK6 (A), SNT15 (B), and MEC04 (C) cells were cultured in medium lacking IL-2 for 48 hours and were then treated with either 0.5 µM or 5 µM of the indicated BH3-mimetic drugs. Cell viability was determined by flow cytometry after staining with annexin V plus PI and data were normalized to DMSO-treated controls or DMSO-treated IL-2 deprived cells as appropriate. (D) Cell viability was determined after treatment with the BCL-XL inhibitor A-1331852 alone or in combination with either the BCL-2 inhibitor ABT-199 or the MCL-1 inhibitor S63845. Cell viability was determined by flow cytometry after staining with annexin V plus PI and data were normalized to DMSO-treated control cells. All compounds were used at a final concentration of 5 µM. Data in panels A and B are presented as means ± SD of 3 independent experiments, each performed in triplicates (n = 3). Significance was determined by the unpaired Student t test and corrected for multiple comparisons using the Holm-Sidak method. *P < .05; **P < .001.
Sensitivity of ENKTL cell lines to the MCL-1 inhibitor S63845 increases upon IL-2 withdrawal or treatment with the BCL-XL inhibitor A-1331852. SNK6 (A), SNT15 (B), and MEC04 (C) cells were cultured in medium lacking IL-2 for 48 hours and were then treated with either 0.5 µM or 5 µM of the indicated BH3-mimetic drugs. Cell viability was determined by flow cytometry after staining with annexin V plus PI and data were normalized to DMSO-treated controls or DMSO-treated IL-2 deprived cells as appropriate. (D) Cell viability was determined after treatment with the BCL-XL inhibitor A-1331852 alone or in combination with either the BCL-2 inhibitor ABT-199 or the MCL-1 inhibitor S63845. Cell viability was determined by flow cytometry after staining with annexin V plus PI and data were normalized to DMSO-treated control cells. All compounds were used at a final concentration of 5 µM. Data in panels A and B are presented as means ± SD of 3 independent experiments, each performed in triplicates (n = 3). Significance was determined by the unpaired Student t test and corrected for multiple comparisons using the Holm-Sidak method. *P < .05; **P < .001.
Cell death induced by BCL-XL–specific BH3-mimetics is enhanced by loss of the MCL-1 gene
To examine the hypothesis that optimal killing of T/NK tumors would be achieved by inhibition of both BCL-XL and MCL-1, CRISPR/Cas9-mediated mutations of MCL-1 were induced in MEC04 cells. Western blot analysis of single cell clones confirmed successful loss of MCL-1 in clones #4, #13, and #22 (Figure 7A). Clones #107, #108, and #109 retained MCL-1 expression as these control cells had instead been transduced with a nontargeting sgRNA. In support of the above hypothesis, MEC04 cells lacking wt MCL-1 could not form tumors in NSG mice (data not shown). Such cells were instead assessed in vitro for survival following treatment with A-1331852, ABT-199 or S63845 (Figure 7B-D). As predicted, MCL-1-depleted MEC04 cells were more sensitive to A-1331852, as the mean IC50 value had decreased 378-fold compared with control cells containing the nontargeting sgRNA (Figure 7E). Perhaps surprisingly, MEC04 cells lacking MCL-1 were sensitized to ABT-199, albeit only at high concentrations. The data further confirm that MCL-1 behaves as the secondary survival factor when BCL-XL action is restricted.

Loss of MCL-1 enhances killing of MEC04 cells by the BCL-XL inhibitor A-1331852 and sensitizes these cells to high doses of the BCL-2 inhibitor ABT199. (A) MEC04 cells stably expressing Cas9 were infected with doxycycline-inducible sgRNAs targeting either human MCL-1 (isgMCL-1_hEx1) or mouse Bcl2l11(Bim) (nontargeting control) (isgNT). Cells were single cell sorted by flow cytometry following doxycycline treatment and then examined for MCL-1 expression by western blot analysis. Dose-response curves generated from annexin V/PI assays following 24-hour treatment with A-1331852 (B), ABT199 (C), or S63845 (D) at the indicated concentrations. (E) IC50 values generated from dose-response curves were grouped by genotype and compared for each drug treatment. Data are presented as means ± SD of 3 independent experiments, each performed in triplicate (n = 3). Significance was determined by the unpaired Student t test and corrected for multiple comparisons using the Holm-Sidak method. **P < .001.
Loss of MCL-1 enhances killing of MEC04 cells by the BCL-XL inhibitor A-1331852 and sensitizes these cells to high doses of the BCL-2 inhibitor ABT199. (A) MEC04 cells stably expressing Cas9 were infected with doxycycline-inducible sgRNAs targeting either human MCL-1 (isgMCL-1_hEx1) or mouse Bcl2l11(Bim) (nontargeting control) (isgNT). Cells were single cell sorted by flow cytometry following doxycycline treatment and then examined for MCL-1 expression by western blot analysis. Dose-response curves generated from annexin V/PI assays following 24-hour treatment with A-1331852 (B), ABT199 (C), or S63845 (D) at the indicated concentrations. (E) IC50 values generated from dose-response curves were grouped by genotype and compared for each drug treatment. Data are presented as means ± SD of 3 independent experiments, each performed in triplicate (n = 3). Significance was determined by the unpaired Student t test and corrected for multiple comparisons using the Holm-Sidak method. **P < .001.
Discussion
ENKTL disease responds poorly to chemotherapy regimens, including CHOP and SMILE,6-8 which initiate apoptosis through DNA damage–induced upregulation of the BH3-only proteins PUMA and NOXA.60 Relapse is common and the median overall survival for patients is only 7.8 months,3 highlighting a need for alternative therapies.
One factor limiting progress for improved therapy is the shortage of accurate cell line models for study. The EBV+ T/NK-cell lines used here were confirmed as representative ENKTL/CAEBV disease models by way of their immunophenotype and EBV gene expression. They are therefore suitable models for testing therapeutic vulnerabilities. The inclusion of both LMP1+ and LMP1− cell lines captured the diversity of EBV gene-expression patterns observed within clinical samples.24 In accordance with the poor effectiveness of the SMILE chemotherapy regimen, all ENKTL/CAEBV cell lines examined exhibited low susceptibility to both etoposide and methotrexate. This resistance could not be correlated to P-glycoprotein activity or the TP53 status, suggesting that other resistance factors were responsible.
We postulated that inducing apoptosis at a point further downstream in the pathway by using novel BH3-mimetic drugs could be an effective therapy for EBV-associated NK-/T-cell tumors. Initially, we anticipated that this therapy may only be effective for those tumors that express LMP1 because LMP1 expression has been shown to upregulate levels of the cellular prosurvival proteins, BCL-2, MCL-1, and BFL-1.25-27 Although BH3-mimetic drugs targeting BCL-2 and MCL-1 were largely ineffective, those that inhibit BCL-XL (A-1331852 and ABT-737) induced substantial cell death in all ENKTL and CAEBV cell lines examined in vitro, irrespective of LMP1 status, indicating a primary survival dependency on BCL-XL. Notably, A-1331852 treatment markedly delayed tumor expansion in an SNK6 xenotransplant mouse model of ENKTL. However, this was not achieved in xenografts of SNT15 or MEC04 cells, and we speculated that this was due to IL-2 deprivation of these malignant cells in NSG mice that do not contain any mature CD4+ T cells, the major source of IL-2.44,61 We therefore predicted that this change in IL-2 availability would alter the expression of key BCL-2 family members. Consistent with this hypothesis, across the cell line panel, the levels of BCL-XL decreased upon IL-2 deprivation and, of note, in SNT15 as well as MEC04 cells, MCL-1 levels increased. This resulted in SNT15 cells becoming less sensitive to the BCL-XL inhibitors and more sensitive to the MCL-1 inhibitor. Although these experiments revealed limitations in in vivo xenotransplant models for investigations into ENKTL, they also gave insights into resistance factors that would come into play when BCL-XL expression was inhibited. Specifically, our results indicate that MCL-1 expression may be a potential resistance factor in patients treated with BCL-XL targeting BH3-mimetics.
In agreement with this, in vitro studies suggested that the most efficacious pairing of BH3-mimetics for the treatment of ENKTL cell lines was BCL-XL inhibition in combination with MCL-1 inhibition. Consistently, in MEC04 cells, the genetic loss of MCL-1 enhanced the efficacy of A-1331852. The reason why SNK6 cells retain sensitivity to A-1331852 in vivo remains unknown but this may be due to the relatively high levels of BCL-XL expression at steady state in these cells. One hypothesis is that the relatively high expression of LMP1 in SNK6 cells could be maintaining sufficient BCL-XL dependency through a strong induction of NF-κB signaling, a pathway known to regulate BCL-XL.62,63
Moving forward, our data suggest that targeting BCL-XL, either directly using BH3-mimetic drugs or indirectly by reducing the expression of BCL-XL through, for example, IL-2 restriction in patients, could be an effective therapeutic strategy for EBV-associated CAEBV and ENKTL. The dependency of platelets on BCL-XL for survival, as was shown in the early clinical trials of navitoclax/ABT-263, which targets BCL-XL, BCL-2, and BCL-W, in patients with lymphoid malignancies or small cell lung cancer,59 will necessitate careful clinical management or targeted delivery of drugs to cancer cells, perhaps by conjugating them to antibodies that bind to the malignant cells. One such antibody-drug conjugate derived from a BCL-XL inhibitor, ABBV-155, is under early clinical investigation as both a monotherapy and in combination with taxane therapy (NCT03595059). Additionally, a platelet-sparing PROTAC degrader of BCL-XL, DT2216, has recently been developed as a better tolerated alternative to navitoclax/ABT-263.64 Our data further indicate that resistance to BCL-XL inhibitors may be mediated by MCL-1 expression, suggesting that dual targeting of both BCL-XL and MCL-1 might provide a more efficacious therapy. Pharmacological inhibition of multiple prosurvival BCL-2 proteins will be challenging due to on-target toxicity to healthy cells but has been achieved for BCL-2 and MCL-1 by using 2 BH3-mimetic drugs in animal models of mantle cell lymphoma, T-cell acute lymphoblastic leukemia, and AML.65-67 In fact, for the treatment of AML, the combination of BCL-2 and MCL-1 inhibition by BH3-mimetics has reached phase 1 clinical trials (NCT03672695). For the combined targeting of BCL-XL and MCL-1 using BH3-mimetics, a tolerable treatment regimen in mouse models has not yet been achieved. One strategy would be to combine BH3-mimetic drugs targeting MCL-1 (which are already in clinical trials), with drugs that target upstream regulators of BCL-XL, such as NF-κB inhibitors.
The lack of efficacy of chemotherapy regimens in ENKTL malignancies, indicated by clinical response data and in the experimental work presented here, mandates that novel options become available. The recent use of BH3-mimetic drugs as effective treatments for various blood cancers suggests that such therapies may also be suitable for patients suffering from an EBV-driven proliferation of T/NK cells.
Data-sharing requests may be e-mailed to the corresponding authors, Gemma L. Kelly and Claire Shannon-Lowe, at gkelly@wehi.edu.au and c.shannonlowe@bham.ac.uk.
Acknowledgments
The authors thank G. Siciliano, H. Johnson, C. Alessandro, K. Hughes, and D. Fayle for mouse husbandry; S. Monard and his team for assistance with flow cytometry; B. Mercer for reviewing karyotypes; and M. Rowe for overall advice on this project.
This work was supported by funding from the Victorian Cancer Agency Fellowship (MCRF 17028) (G.L.K.), Cancer Council Victoria, grants-in-aid #1086157 and #1147328 (G.L.K.); the National Health and Medical Research Council (NHMRC), project grant #1086291 (G.L.K.), program grant #1113133 (A.S.), fellowship #1020363 (A.S.); the Leukaemia Foundation Australia grant (G.L.K. and A.S.); the Leukemia & Lymphoma Society grant #7015-18 (A.S.); the estate of Anthony (Toni) Redstone OAM, The Craig Perkins Cancer Research Foundation, and The Dyson Bequest; operational infrastructure grants through the Australian Government NHMRC Independent Research Institutes Infrastructure Support Scheme (IRIISS) and the Victorian State Government Operational Infrastructure Support; a Universitas 21 project grant (C.S.-L., H.M.L., and G.L.K.); and the Medical Research Council, Research Grant #MR/N023781/1 (C.S.-L.)
Authorship
Contribution: N.S., L.C.G., R.J.T., C.C., O.K., R.N.M., P.L., and C.S.-L. performed the experiments; G.L., H.M.L., A.S., C.S.-L., and G.L.K. provided resource; N.S., H.M.L., A.S., C.S.-L., and G.L.K. contributed to the conception and design of the research; N.S., R.J.T., O.K., and R.N.M. analyzed the data; N.S., R.J.T., R.N.M., A.I.B., H.M.L., A.S., C.S.-L., and G.L.K. interpreted the data; N.S. and G.L.K. wrote the manuscript; and N.S., A.S., C.S.-L., and G.L.K. edited and critically revised the manuscript.
Conflict-of-interest disclosure: The Walter and Eliza Hall Institute receives milestone and royalty payments related to venetoclax, and employees (N.S., C.C., O.K., P.L., G.L., A.S., and G.L.K.) may be eligible for benefits related to these payments. The laboratory of A.S. receives research funding from Servier. The remaining authors declare no competing financial interests.
Correspondence: Gemma L. Kelly, The Walter and Eliza Hall Institute for Medical Research, 1G Royal Parade, Parkville, Melbourne, VIC 3052, Australia; e-mail: gkelly@wehi.edu.au; and Claire Shannon-Lowe, Institute of Immunology and Immunotherapy, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, United Kingdom; e-mail: c.shannonlowe@bham.ac.uk.