

Key Points
A novel nonmyeloablative allotransplant is safe and effective in cutaneous T-cell lymphoma.
Cutaneous T-cell lymphoma patients achieved clinical and molecular remission after allotransplant.

Abstract
The majority of patients with refractory, advanced-stage mycosis fungoides (MF) or Sézary syndrome (SS) have a life expectancy of <5 years. Here, we report a phase 2 study of a novel nonmyeloablative allogeneic transplantation strategy tailored for this patient population. This study has completed the enrollment, and 35 patients (13 MF, 22 SS) have undergone transplant as planned. The majority (80%) of the patients had stage IV disease and received multiple previous systemic therapies. All patients had active disease at the time of conditioning using total skin electron beam therapy, total lymphoid irradiation, and antithymocyte globulin, and received allograft infusion as outpatients. Cyclosporine or tacrolimus and mycophenolate mofetil were used for graft-versus-host disease (GVHD) prophylaxis. Patients tolerated the transplant well, with 1- and 2-year nonrelapse mortality of 3% and 14%, respectively. The day +180 cumulative incidence of grade 2 to 4 acute GVHD was 16%, and the 2-year incidence of moderate/severe chronic GVHD was 32%. With a median posttransplant follow-up of 5.4 years, the 2-, 3-, and 5-year overall survival rates were 68%, 62%, and 56%. Using high-throughput sequencing of the T-cell receptor for minimal residual disease monitoring, we observed that 43% achieved molecular remission, which was associated with a lower incidence of disease progression or relapse (9% vs 87%; P = .02). Our study also showed that patients who were aged ≥65 years at the time of allotransplant had similar clinical outcomes compared with younger patients. Thus, we have developed an alternative and potentially curative nonmyeloablative allogeneic transplant regimen for patients with advanced stage MF/SS. This trial was registered at www.clinicaltrials.gov as #NCT00896493.
Introduction
Mycosis fungoides (MF) and Sézary syndrome (SS) are the most common type of cutaneous T-cell lymphoma.1,2 Treatment selection is based on clinical stage, age, comorbidities, and presence of adverse factors such as large cell transformation.3 Although new systemic therapies such as histone deacetylase inhibitors (eg, vorinostat, romidepsin), pralatrexate, brentuximab vedotin, and mogamulizumab are available, the response rates vary, and the duration of response is often limited.4-10 Total skin electron beam therapy (TSEBT) can be used effectively to optimize clearing of skin disease.11-13 Patients with advanced-stage MF/SS are often treated with sequential therapies; they eventually become refractory to available agents and have a median survival of <5 years.14,15
In contrast to other forms of lymphoma, malignant T cells in MF/SS are not as sensitive to cytotoxic chemotherapies.16 Therefore, it is not unexpected that high-dose chemotherapy followed by autologous hematopoietic cell transplant has not shown durable clinical benefit.17 Several recent reports have described clinical efficacy using allogeneic transplant.18-23 This approach relies primarily on immunologic graft-versus-lymphoma (GVL) effects to eradicate residual lymphoma cells after cytoreduction. However, allotransplantation-associated toxicity and complications have been major barriers to its application, especially for older patients and those with comorbid medical conditions. A major concern in the management of posttransplant patients with MF/SS is infectious complications, partly due to a suppressed immune system and breakdown of the skin barrier.24 Indeed, severe and fatal posttransplant infections have been reported.18-23 In addition, graft-versus-host disease (GVHD) is a major potential complication, with a reported incidence of acute GVHD of 40% to 76% and chronic GVHD of 35% to 83%. Although reduced-intensity conditioning has decreased these transplant-related complications, most conditioning regimens are still associated with high 1-year nonrelapse mortality (NRM) of 11% to 25%.18-23
To overcome the problems associated with currently available regimens, we developed a novel nonmyeloablative conditioning consisting of TSEBT, total lymphoid irradiation, and antithymocyte globulin (TSEBT-TLI-ATG). Here we report the outcomes from a phase 2 clinical trial in patients with advanced-stage MF/SS, with additional focus on the utilization of high-throughput sequencing (HTS) to monitor minimal residual disease (MRD).
Methods
Patient population
The cohort consisted of the 35 patients with MF/SS who enrolled in a prospective phase 2 clinical trial approved by the Institutional Review Board (ClinicalTrials.gov #NCT00896493). All patients provided informed consent in accordance with the Declaration of Helsinki and enrolled on this trial before starting the conditioning regimen with TSEBT-TLI-ATG. Eligibility criteria included age ≥18 years without an upper age limit, stage IIB to IV disease, and failure of at least one standard systemic therapy. Patients with pulmonary diffusion capacity <40%, cardiac ejection fraction <30%, Karnofsky performance status <70%, history of HIV infection, and who were pregnant were excluded. Donors and recipients underwent high-resolution HLA typing for class I (-A, -B, and -Cw) and class II (-DRB1 and -DQB1) molecules.
Transplant regimen
The conditioning regimen consisted of TSEBT, TLI, and ATG. Using the Stanford 6-dual field technique,25 TSEBT was initiated 6 to 9 weeks before beginning TLI-ATG. The radiation dose was adjusted according to previous radiation treatment and disease status with a target dose of 30 to 36 Gy. The last 2 weeks of TSEBT overlapped with TLI-ATG in most of the cases. TLI-ATG was administered as previously described.26,27 In brief, rabbit ATG (Thymoglobulin; Genzyme, Cambridge, MA) was infused at 1.5 mg/kg per day for 5 consecutive days, beginning on day −11. TLI was administered at a dose of 0.8 or 1.2 Gy/d from day −11 through day −7, and from day −4 through day −1, with an additional fraction delivered in the afternoon of day −1 for a total dose of 8 or 12 Gy. The first 3 patients received 12 Gy, whereas the remainder received 8 Gy TLI after protocol modification. The total dose of TLI was then changed back to 8 Gy as in the original TLI-ATG study for uncertain advantage for engraftment with 12 Gy and potential benefit of less cytopenia with 8 Gy after observing suboptimal engraftment in 2 of the first 3 patients (1 mixed donor cell chimerism and 1 graft failure).26,27 All patients received unmanipulated granulocyte colony-stimulating factor–mobilized peripheral blood hematopoietic cells on day 0. GVHD prophylaxis consisted of cyclosporine or tacrolimus and mycophenolate mofetil as previously described.27 The entire regimen required only a 5-day inpatient stay during the ATG infusion.
Donor chimerism and T-cell receptor high-throughput sequencing
DNA genotyping of polymorphic markers encoding short tandem repeats was used to quantitate donor chimerism.28 Full donor chimerism was defined as the achievement of at least 95% donor CD3+ chimerism in the blood at any time point. Graft failure was defined as failure to surpass 5% donor CD3+ chimerism,27 whereas mixed chimerism was defined as donor CD3+ chimerism ranging from 5% to 94%.29 The monitoring of MRD using T-cell receptor high-throughput sequencing (TCR HTS) in blood and skin biopsy samples has been described previously.30,31 Diagnostic blood and skin samples were used to identify malignant clones. Blood and skin samples were taken before TSEBT as baseline and subsequently posttransplant at day +30, +60, +90, +180, +270, +360, and then yearly. One or more posttransplant skin samples were obtained at known pretransplant disease sites or from suspicious skin lesions.
End points and statistical analysis
Disease status at the time of conditioning, global clinical responses, and disease progression were defined according to criteria of the MF/SS consensus guidelines.32 Progression-free survival (PFS) was calculated from the date of transplantation to disease progression or death of any cause. Event-free survival (EFS) was calculated from the date of transplantation to disease progression, relapse in those who achieved a complete response (CR), initiation of lymphoma-specific therapy (excluding topical steroid), or death of any cause, whichever occurred first with only the last follow-up censored. This was a stricter end point than the PFS by the MF/SS consensus guidelines. The graft failure was not considered an event in EFS nor censored in either PFS or EFS. GVHD was graded by using current consensus criteria.33,34 The cumulative incidence of GVHD was estimated by using the Kaplan-Meier method in which graft failure and death were censored.
The study planned to enroll 35 patients based on feasibility for completion in a timely fashion. The primary efficacy end point was PFS at day +180 with a target rate of 75% and a 95% confidence interval (CI) half-width of 15%. The secondary end points included EFS, overall survival (OS), and NRM at 2 years’ posttransplant. PFS, EFS, OS, and time to GVHD were analyzed by using the Kaplan-Meier method. The cumulative incidence of progression/relapse was estimated by using the Cox proportional hazards model in which death unrelated to disease was treated as competing risk events and last follow-up were censored. The cumulative incidence of progression/relapse was compared between patients with molecular remission vs without by using the Wald χ2 test. All tests were 2-sided, and P < .05 was considered significant. All statistical analyses were performed by using SAS version 9.4 (SAS Institute, Inc., Cary, NC).
Results
Transplantation and engraftment
The study completed the enrollment of 35 patients as planned. The study cohort was composed of all the consecutive patients who received allogeneic transplant in our institute during this period of time. The baseline characteristics of the 35 patients are described in Table 1. Thirteen patients (37%) had MF, and 22 patients had SS. This cohort represented a heavily treated, refractory group of patients with median of 5 previous systemic therapies. All but one patient with MF had recently documented large cell transformation (LCT), an adverse prognostic factor, in the skin and four patients with additional LCT in extracutaneous sites (3, lymph node; 1, tonsil). Eight patients with SS had LCT in either the skin or lymph nodes. Although no patient had progressive disease at the time of the conditioning, none was in CR (23 PR, 12 stable disease) from the last systemic therapy before TSEBT. All patients, except one who had experienced a severe skin reaction from previous TSEBT, received TSEBT with median dose of 32 Gy (range, 20-36 Gy). The median dose of TSEBT was 36 Gy (range, 20-36 Gy) for patients with MF and 28 Gy (range, 23-36 Gy) for patients with SS. Thirteen patients had HLA-identical sibling donors, and the remainder had unrelated donors. As permitted by the protocol, 2 patients had the TSEBT interrupted to treat their extracutaneous disease (1 in blood and lymph node, 1 in tonsil) with denileukin diftitox and brentuximab vedotin, respectively. An additional 2 patients received extracorporeal photopheresis, and 1 patient received a single low dose of alemtuzumab during TSEBT to stabilize Sézary cell burden in the blood.
The median donor CD34+ cell dose was 6.8 (range, 1.6-12.4) × 106 cells/kg, and the median donor CD3+ cell dose was 252 (range, 127-631) × 106 cells/kg. At day +90, 15 patients (43%) achieved full donor chimerism, and 16 patients (46%) had mixed chimerism. Four patients (11%) had primary graft failure. Of the 16 patients with mixed chimerism at day +90, a total of 7 patients subsequently achieved full donor chimerism, 4 had persistent mixed chimerism, and 5 had secondary graft failure. The median time to achieving full donor chimerism was 56 days (range, 28-270 days). There was no difference in the donor CD34+ or CD3+ cell dose between patients achieving full donor chimerism, mixed chimerism, and graft failure (P = .21 and P = .08, respectively). All patients with graft failure recovered autologous hematopoiesis.
Immediate posttransplant toxicity
All patients received their hematopoietic cell infusions as outpatients. Twenty-two patients required hospital admission in the first 100 days posttransplant, with a median length of hospital stay of 4 days (Table 2). All admitted patients were treated successfully and discharged, with the exception of 1 patient who died on day +103 due to gastrointestinal (GI) acute GVHD. Six (17%) patients had posttransplant neutropenia (absolute neutrophil count <0.5 × 109/L), and 2 (6%) patients had severe thrombocytopenia (platelet count <10 × 109/L) during the first 100 days.
Seventeen (63%) of the 27 patients at risk for cytomegalovirus (CMV) reactivation developed CMV viremia, requiring preemptive therapy without any patient developing CMV organ disease. Epstein-Barr virus reactivation was seen in 10 patients (29%), including 2 cases of documented posttransplant lymphoproliferative disease, both of which resolved with rituximab therapy. Four patients had possible central venous catheter–related infections, including 1 case of documented bacteremia; all were treated with intravenous antibiotics to resolution.
Graft-versus-host disease
Four patients developed grade 1 acute GVHD (all skin). Four patients had grade 2 acute GVHD (skin, skin/GI, GI, liver), and 1 patient had grade 4 GI acute GVHD that contributed to his death. The median time to onset of acute GVHD was 60 days (range, 25-154 days). Two patients with grade 2 acute GVHD (GI, liver) were late-onset beyond day +100. One of these two patients experienced morning sickness from an unplanned pregnancy and failed to adhere to the scheduled immunosuppressive regimen before the development of GVHD. The estimated incidence of grade 2 to 4 and 3 to 4 acute GVHD at day +180 was 16% (95% CI, 2-49) and 3% (95% CI, 0-61), respectively (Figure 1A).
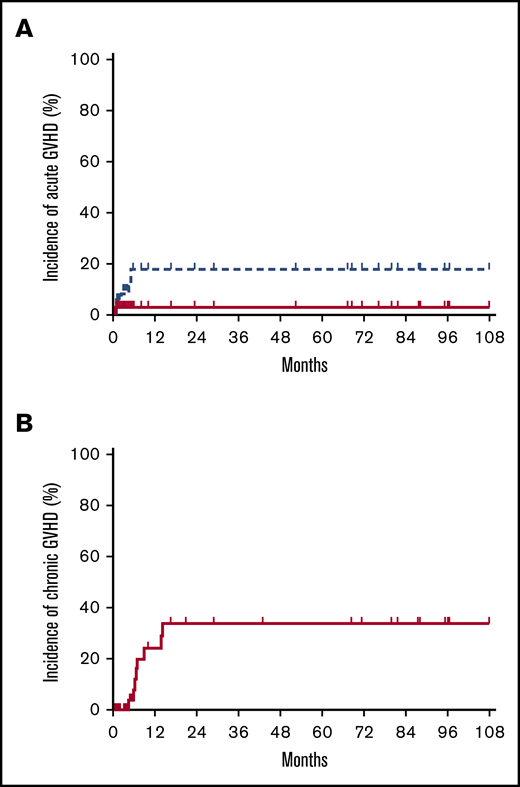
Cumulative incidences of GVHD. Cumulative incidences of acute GVHD grades 2 to 4 (blue dotted line in A), grade 3 to 4 (red solid line in A), and moderate/severe chronic GVHD (B).
Cumulative incidences of GVHD. Cumulative incidences of acute GVHD grades 2 to 4 (blue dotted line in A), grade 3 to 4 (red solid line in A), and moderate/severe chronic GVHD (B).
Five patients had progression of acute GVHD to chronic GVHD: one with mild skin involvement, two with severe skin involvement, and two with multi-organ involvement. Four additional patients developed de novo moderate/severe chronic GVHD without a history of acute GVHD. The median time to onset of chronic GVHD was 193 days (range, 131-424 days). The estimated incidence of moderate/severe chronic GVHD at 2 years was 32% (95% CI, 11-58) (Figure 1B). There was no difference in incidence of grade 2 to 4 acute GVHD or moderate/severe chronic GVHD between patients with a sibling vs unrelated donor graft (both, P = 1.00).
Clinical response and survival
The median follow-up after transplant was 5.4 years (range, 0.3-10.4 years) for the entire group. At day +90, 20 patients (57%) achieved a global CR, 8 had a partial response (PR; overall response rate, 80%), and 3 had stable disease. Four patients experienced progressive disease within 90 days after transplant. A majority of the patients with SS (73%, n = 16) achieved CR, including 4 of the 8 patients with LCT. Patients with MF had a CR rate of 31%. In the entire group, the CR rate was higher among those patients who had no evidence of LCT compared with those who had LCT (80% vs 40%, respectively; P = .037). Risk stratification using the Retrospective Cutaneous Lymphoma International Prognostic Index (Retro-CLIPI)35 is shown in Table 1. The overall response rate was 78% for the high-risk group (11 CR, 3 PR, 1 stable disease, and 3 progressive disease), 85% for the intermediate-risk group (8 CR, 3 PR, 1 stable disease, and 1 progressive disease), and 75% for the low-risk group (1 CR, 2 PR, and 1 stable disease).
As the specified primary end point, the day +180 PFS was estimated to be 73% (95% CI, 55-85). The 2-year EFS and OS were estimated to be 40% (95% CI, 24-56) and 68% (95% CI, 50-81), respectively. With a median follow-up of 5.4 years, 5-year EFS, PFS, and OS were 26% (95% CI, 13-41), 41% (95% CI, 24-58), and 56% (95% CI, 38-71) (Figure 2). The 5-year OS was similar between patients who had no LCT vs those who had LCT (58% vs 53%; P = .490). The 5-year OS was 50%, 62%, and 67% for the Retro-CLIPI high-, intermediate-, and low-risk groups (P = .759). The 5-year OS was similar between patients who were aged ≥65 years and those who were aged <65 years (50% vs 60%; P = .791) or who were aged <60 years (50% vs 51%; P = .709) at time of transplant. The 5-year EFS, PFS, and OS for patients with graft failure were 11% (95% CI, 1-49), 20% (95% CI, 1-58), and 40% (95% CI, 18-79).
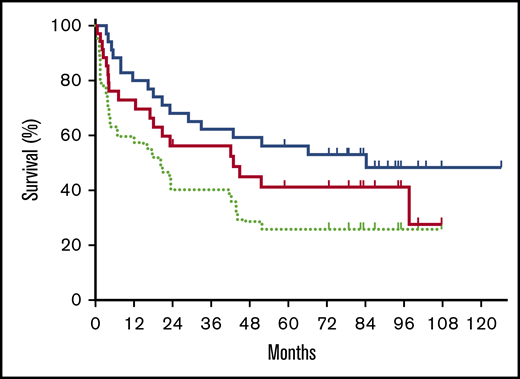
Survival posttransplant. Kaplan-Meier estimates of OS (blue solid line), PFS (red solid line), and EFS (light green dotted line).
Survival posttransplant. Kaplan-Meier estimates of OS (blue solid line), PFS (red solid line), and EFS (light green dotted line).
Twenty patients experienced disease progression or relapse after transplant with only 1 patient without skin involvement (skin in 19 of 20, blood in 7 of 20, lymph node in 7 of 20, 1 in liver, and 1 in lung). All 20 patients received treatments, including topical steroid in most of the patients, local radiation (40%), TSEBT (20%), conventional chemotherapy including antibody-drug conjugates (80%), histone deacetylase inhibitor (35%), systemic immunomodulation (30%), donor lymphocyte infusion (DLI) (40%), and novel agent(s) under clinical trial (25%); the median number of systemic therapies was 3 (range, 1-10). Of the 8 patients who had DLI, 3 achieved CR, 2 achieved PR, 2 had stable disease, and 1 had progressive disease. Six of the 9 patients with graft failure received a second allogeneic transplant, with 3 patients achieving long-term disease control thereafter.
Of the 17 deaths in the study, 5 were due to NRM. The 1- and 2-year cumulative incidence of NRM were 3% (95% CI, 0-8) and 14% (95% CI, 2-27), respectively. Causes of NRM included acute GVHD, chronic GVHD, secondary malignancy (pre-B acute lymphoblastic leukemia), hepatitis B, and hemorrhage secondary to anticoagulation in 1 patient each (Table 2).
Molecular response
Thirty patients had diagnostic samples available for HTS to determine the unique TCR CDR3 sequences from the malignant clones. Immediately before conditioning with TSEBT, 28 patients (93%) had detectable disease by HTS in blood (<1%, 13 patients; 1%-5%, 4 patients; and >5%, 11 patients). After transplant, 13 (43%) of the 30 patients achieved sustained molecular remission in blood (malignant clone undetectable with a sensitivity of 1 in 200 000 nuclear cells) with a median time to remission of 146 days (range, 30-730). Patients with full donor chimerism (n = 20) had a higher chance of achieving molecular remission in blood than those with mixed donor chimerism (65% vs 0%; P < .0001). Although none of the patients with graft failure achieved sustained molecular remission, 1 patient had a transient molecular remission and 2 other patients had reduction of MRD posttransplantation. Of the 13 patients who achieved molecular remission in blood, 8 achieved concurrent molecular remission in the skin. Of the remaining 5 patients, 3 subsequently achieved molecular remission in the skin (∼4 more months later), whereas 2 patients had persistent MRD in the skin. The 10 patients who achieved complete clinical response but had persistent MRD all had a very low tumor burden according to HTS (median MRD in blood, 0.101% [range, 0.002%-1.532%]; median MRD in skin, 0.400% [range, 0.035%-1.508%]). As the entire group, the estimated cumulative incidence of disease progression or relapse after transplant was 59% (95% CI, 45-77) at 5 years. Patients who had MRD according to HTS either in blood or skin had higher chance of disease progression or relapse than patients who achieved molecular remission in both blood and skin, with a cumulative incidence of progression or relapse of 87% and 9%, respectively (P =.002) (Figure 3A). In addition, within the 18 patients who achieved complete clinical remission after transplant, MRD detected by HTS was also associated with a higher cumulative incidence of progression or relapse than those without MRD (75% vs 13%; P = .028) (Figure 3B).
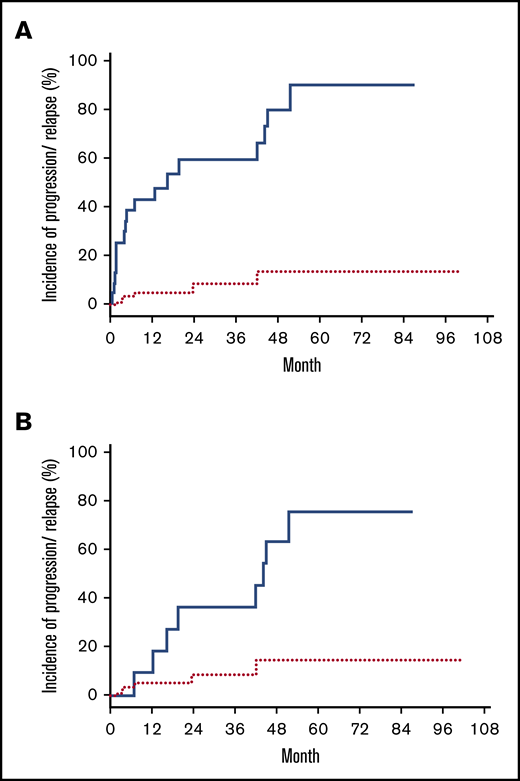
Incidence of disease progression/relapse posttransplant. Cumulative incidence of disease progression/relapse in patients who achieve molecular remission (red dotted line) and in patients who had MRD (blue solid line). (A) All patients. (B) Patients in CR.
Incidence of disease progression/relapse posttransplant. Cumulative incidence of disease progression/relapse in patients who achieve molecular remission (red dotted line) and in patients who had MRD (blue solid line). (A) All patients. (B) Patients in CR.
Discussion
In this prospective phase 2 clinical trial of heavily treated, refractory patients with advanced-stage MF (majority with LCT) or SS, we observed a high overall response rate, including a CR rate of 57%. For the 15 patients who failed to achieve CR at day +90 posttransplant, all had cutaneous disease, including 8 patients who had only residual skin lesions with resolution of pretransplant extracutaneous disease. It is likely that the GVL effects were very effective against extracutaneous diseases, especially the blood compartment. Consistent with this notion, patients with SS had a significantly higher CR rate than patients with MF in our study. Furthermore, our unique conditioning with TSEBT-TLI-ATG may have significant clinical benefit in reducing the disease burden in the skin, lymph node, and blood, respectively. Because all patients had active skin disease at the time of conditioning, the maximally tolerated dose of TSEBT was used in all but 1 patient. Although TSEBT is very effective in controlling disease in the skin,11,12 the full benefit of the conditioning TSEBT is difficult to assess given its proximity to allograft infusion. Two of the 3 patients who had high Sézary cell burden measured by HTS before ATG infusion showed significant decreases in the circulating Sézary cells 1 week after ATG on the day of allograft infusion (from 68% to 11%, 40% to 1.5%, and 29% to 21%, respectively), suggesting a cytoreductive effect in blood by ATG. Thus, our TSEBT-TLI-ATG regimen uniquely provides a highly effective disease-specific cytoreduction in addition to the GVL activity. On the other hand, GVL activity is critical for long-term disease control. This is evident by a significantly higher incidence of disease progression or relapse in patients who did not achieve full donor cell chimerism (92% vs 46%; P = .005) or who experienced graft loss (89% vs 57%; P = .014). In addition, in 4 patients whose Sézary cell burden was measured on the day of allograft infusion, all exhibited a significant reduction of disease burden by day +30 as assessed by HTS (from 11% to 0.9%, 1.5% to 0%, 21% to 8.8%, and 27% to 0.2%).
Currently, there is no consensus on who should be considered for allogeneic transplantation or the timing of transplantation in the course of their disease. In general, transplant has been reserved for patients with advanced-stage disease after failing several lines of systemic therapies or earlier in those with highly aggressive disease. At Stanford, the decision for allogeneic transplant was made jointly by the members of a multidisciplinary cutaneous lymphoma clinic, including dermatology, medical oncology, radiation oncology, and transplantation specialists considering patients with aggressive clinical course, failure of multiple therapies (median of 5), and with expected survival <5 years without transplant. All patients who received allogeneic transplant during this period of time were enrolled in this study after adequate control of their disease and finding of a suitable donor.
Availability of new systemic therapies, including pralatrexate, brentuximab vedotin, and mogamulizumab, has provided more treatment options for patients with advanced-stage MF/SS in recent years. This has contributed to the decreased number of patients enrolled at the later part of this study because we were able to use these newer therapies to delay the timing of transplant in some patients. Although the clinical outcomes in phase 2 studies are greatly affected by patient selection, patients in this clinical trial had mostly advanced stage 4 (80%) disease, LCT (57%), high/intermediate Retro-CLIPI risk (89%), had failed at least 2 systemic therapies, and were older (median age >60 years). Thus, our study included a patient population with poor prognosis without transplant. The OS in the high- and intermediate-risk groups in this study compared favorably with OS in the similar risk groups in the Retro-CLIPI cohort, in which the OS was calculated from time of diagnosis (5-year OS in the high-risk group, 50% vs 28%; 5-year OS in the intermediate-risk group, 62% vs 44%). The results suggest a clinical benefit of allogeneic transplantation in these 2 risk groups. In addition, the tolerability and low NRM (3% in 1 year) of this regimen has enabled us to safely perform allotransplantation in an older patient population compared with other reports (median, 62 years vs 44-53 years) (Table 3).18-23 Twenty patients (57%) in this study were older than 60 years at the time of transplant (60-64 years old, 8 patients; 65-69 years old, 6 patients; and >70 years old, 6 patients). In this study, age had no impact on the clinical outcomes, with patients who were ≥65 years old having similar OS compared with that of younger patients.
The incidence of grade 2 to 4 acute GVHD and chronic GVHD in our study seemed to be significantly lower than reported previously by other groups (16% vs 28% to 49% and 32% vs 35% to 83%, respectively).18-23 Interestingly, 57% of patients who developed acute GVHD and all patients with chronic GVHD had skin manifestations of GVHD. Although it is possible that the addition of TSEBT induced cutaneous inflammation and predisposed patients for skin GVHD by affecting host skin dendritic cells,36 the overall incidence of skin acute GVHD (all grades, 14%) in this study was significantly lower than the 57% to 59% incidence reported previously for similar patients.18,21,23
Two-thirds of the patients in our study received unrelated donor grafts, including 7 patients with HLA-mismatched donors. In contrast to the French study,21 we found no difference in the incidence of GVHD or OS between sibling and unrelated donors. However, we observed a higher rate of graft failure than reported in other studies of MF/SS,19-21 or in other patient populations who received TLI-ATG conditioning.26,27,37 Eight cases of graft failure occurred in unrelated donors, including 4 with mismatched donors and 1 in an HLA-matched related donor. Four patients experienced graft failure in the setting of persistent or progressive disease, which is consistent with the experience in other hematopoietic malignancies.38-40 Compared with patients with other lymphomas who underwent TLI-ATG allotransplantation, patients with MF/SS had fewer cytotoxic therapies immediately before allotransplant. Thus, the relatively preserved host immunity may be contributing to the higher risk of graft failure.41 In contrast to other reports,42 we detected no measurable anti-HLA antibodies against donor cells before transplantation or after graft failure.
By using TCR HTS, we were able to monitor MRD in a quantitative manner with high specificity and sensitivity.30 The immediate posttransplant GVL effect was observed even without full donor engraftment, followed by sustained disease control that may depend on full donor engraftment. Given the large number of donor T cells (range, 129-631 × 106 cells/kg) infused along with hematopoietic precursor cells on day 0, it is possible that these donor T cells were responsible for the immediate GVL effect. Nearly one-half of the patients achieved molecular remission in blood, 6 occurring >6 months posttransplant. This delayed molecular remission is consistent with our experience in patients with chronic lymphocytic leukemia using TLI-ATG conditioning.43 Most patients (85%) who achieved molecular remission in blood also achieved molecular remission in the skin. Although the number of patients in this study was relatively small, our results suggest a significant clinical benefit among those who attain molecular remission (Figure 3). A study with a larger number of patients is needed to confirm the clinical significance of achieving molecular remission measured by using HTS.
The management of disease progression after allogeneic transplantation can be challenging and must be individualized to address the clinical extent and severity of each patient’s disease. The general strategies have always been reduction of disease burden and enhancement of the GVL effect. The former can be achieved by skin-directed and/or systemic therapies in a similar escalating manner as in nontransplant patients. There are several ways to enhance the GVL effect, including withdrawal of immunosuppression, administration of DLI, or nonspecific immunomodulation (eg, low-dose interferon). At Stanford, patients with disease progression or relapse were managed jointly at the multidisciplinary cutaneous lymphoma clinic. Although these patients received skin-directed management, all patients received systemic therapies, including chemotherapy, immunomodulator, DLI, and second allogeneic transplant in patients who had graft failure. The choice of systemic therapy depended on patient’s disease status (skin vs extracutaneous; aggressiveness) and previous response to specific agent(s). The goal is to reduce the disease burden, followed by enhancement of GVL effect by different means. In contrast to a previous report,22 DLI has not provided consistent clinical benefit in our study. Although our regimen is safer compared with other transplant strategies, the major disadvantage is the slower and sometimes incomplete engraftment, including higher rate of graft rejection leading to early disease progression or relapse. To mitigate this scenario, strategies to improve the speed and the completeness of engraftment using donor cell chimerism–driven early donor CD8+ memory T lymphocyte infusion (clinicaltrials.gov #NCT02424968) or replacing the last dose of TLI with a very low dose total body irradiation (TBI, 0.8 Gy) in standard TLI-ATG (TLI-ATG-TBI) conditioning (clinicaltrials.gov #NCT03734601) are currently under investigation. The TLI-ATG-TBI conditioning seems to confirm improvement in the rate of day +30 full donor cell engraftment and does not increase the incidence of GVHD in treating patients with other hematologic malignancies (W.K.W. and R.L., unpublished observation, June 2020).
Similar to a previous report,18 we also observed a prompt clinical response in patients with SS, compared with patients with MF. In addition, patients with refractory tumor lesions before the conditioning regimen tend to have posttransplant relapse in the skin compartment. However, the 5-year survival rates were not different between patients with MF and patients with SS. Therefore, this unique TSEBT-TLI-ATG conditioning should be an appropriate transplant option for patients with SS or MF when optimal disease control of skin lesions is achieved.
In conclusion, we showed that TSEBT-TLI-ATG conditioning followed by allogeneic transplantation is safe and well tolerated in patients with advanced-stage MF and SS, even in an older patient population. This novel incorporation of TSEBT with TLI-ATG conditioning provided unique benefit to patients with MF/SS because TSEBT provided consolidative disease control in the skin while TLI-ATG conditioning had low GVHD and NRM. Nearly one-half of the patients assessed for MRD achieved a molecular remission, which was associated with longer duration of clinical remission. Future study is needed to increase the efficacy of this transplant strategy by facilitating the donor engraftment and overcoming the high rate of graft rejection in this patient population.
Part of this work was presented at the 56th annual meeting of the American Society of Hematology, San Francisco, CA, 6-9 December 2014.
Requests for data sharing may be submitted to the corresponding author (Wen-Kai Weng; e-mail: wkweng@stanford.edu).
Acknowledgments
This work was supported by the Haas Family Foundation, and Albert Yu and Mary Beckmann Foundation. W.-K.W. is the recipient of a Stanford University Cancer Institute Developmental Cancer Research Award and Stanford Institute for Immunology, Transplantation and Infection Seed Grant Award. A.R. is a recipient of an American Cancer Society Mentored Scholar Research Grant.
Authorship
Contribution: W.-K.W. and Y.H.K. designed research, performed research, collected data, analyzed and interpreted data, and wrote the manuscript; R.T.H. designed research, analyzed and interpreted data, and wrote the manuscript; S.A., A.R., L.J., R.L., D.M., J.S., L. Muffly, E.M., R.S.N., and L. Million performed research and analyzed and interpreted data; E.W. and T.A. collected data, and analyzed and interpreted data; M.K. recruited patients, and analyzed and interpreted data; and S.L. analyzed and performed statistical analysis.
Conflict-of-interest disclosure: A.R. received research funding from Pharmacyclics. D.M. served on the advisory board for Adaptive Biotechnologies, AlloGene, Janssen, Juno-Celgene-BMS, Kite-Gilead, Miltenyi Biotec, Novartis, Pharmacyclics, and Precision Bioscience; and received research funding from Adaptive Biotechnologies, Becton Dickinson, Genentech, Isolexis, Kite-Gilead, Novartis, and Pharmacyclics. R.S.N. consulted for Amgen, Kuur, Magenta Therapeutics, and Jazz; and owns stocks of Magenta Therapeutics and BioEclipse Therapeutics. M.K. consulted for Kyowa-Kirin and Seattle Genetics. Y.H.K. served on the advisory board for Galderma, Innate, Kyocera, Corvus, and Seattle Genetics; and received research funding from Corvus, Eisai, Elorac, Galderma, Innate, Kyowa Kirin, Portola, Soligenix, and Trillium Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Wen-Kai Weng, Division of Blood & Marrow Transplantation, Department of Medicine, Stanford University School of Medicine, 300 Pasteur Dr, H0101, MC 5623, Stanford, CA 94305; e-mail: wkweng@stanford.edu.



