Key Points
Mature data from large randomized trial shows that consolidative HDT/ASCT after frontline R-CHOP for FL does not improve OS.
Genotype-based risk models do not identify patient subsets with OS benefit from frontline HDT/ASCT.

Abstract
High-dose therapy and autologous stem cell transplantation (HDT/ASCT) is an effective salvage treatment for eligible patients with follicular lymphoma (FL) and early progression of disease (POD). Since the introduction of rituximab, HDT/ASCT is no longer recommended in first remission. We here explored whether consolidative HDT/ASCT improved survival in defined subgroups of previously untreated patients. We report survival analyses of 431 patients who received frontline rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for advanced FL, and were randomized to receive consolidative HDT/ASCT. We performed targeted genotyping of 157 diagnostic biopsies, and calculated genotype-based risk scores. HDT/ASCT improved failure-free survival (FFS; hazard ratio [HR], 0.8, P = .07; as-treated: HR, 0.7, P = .04), but not overall survival (OS; HR, 1.3, P = .27; as-treated: HR, 1.4, P = .13). High-risk cohorts identified by FL International Prognostic Index (FLIPI), and the clinicogenetic risk models m7-FLIPI and POD within 24 months–prognostic index (POD24-PI) comprised 27%, 18%, and 22% of patients. HDT/ASCT did not significantly prolong FFS in high-risk patients as defined by FLIPI (HR, 0.9; P = .56), m7-FLIPI (HR, 0.9; P = .91), and POD24-PI (HR, 0.8; P = .60). Similarly, OS was not significantly improved. Finally, we used a machine-learning approach to predict benefit from HDT/ASCT by genotypes. Patients predicted to benefit from HDT/ASCT had longer FFS with HDT/ASCT (HR, 0.4; P = .03), but OS did not reach statistical significance. Thus, consolidative HDT/ASCT after frontline R-CHOP did not improve OS in unselected FL patients and subgroups selected by genotype-based risk models.
Introduction
Follicular lymphoma (FL) is one of the most common subtypes of non-Hodgkin lymphomas. It is a well-defined but heterogeneous clinicopathologic entity.1 Most patients are diagnosed with advanced disease and cannot be cured with conventional therapies. FL with histologic grades 1, 2, and 3a is considered the prototype non-Hodgkin lymphoma entity with indolent clinical course. However, even with modern immunochemotherapies, ∼20% of patients experience early progression of disease (POD) (ie, POD within 24 months [POD24]) and have a median overall survival (OS) of <5 years.2-4
Retrospective studies have demonstrated prolonged survival in patients with POD24 who received high-dose therapy (HDT) followed by autologous stem cell transplantation (ASCT).5-7 Accordingly, US and European guidelines currently recommend offering HDT/ASCT to eligible patients (ie, patients with POD24 who are medically fit and have achieved a second remission).8,9
We have recently shown that the clinicogenetic risk models m7-FLIPI and POD24 prognostic index (POD24-PI), both integrating somatic gene mutation status and the FL International Prognostic Index (FLIPI) improve pretreatment risk stratification10 and can identify a high-risk cohort of patients who have an increased risk of developing POD24 before initiation of frontline treatment.3 Although consolidative HDT/ASCT is no longer recommended in patients in first remission after state-of-art immunochemotherapy, we sought to explore if it is possible to identify a subgroup of previously untreated patients with high-risk profiles who may have a survival benefit from dose-intensified frontline regimens.
Methods
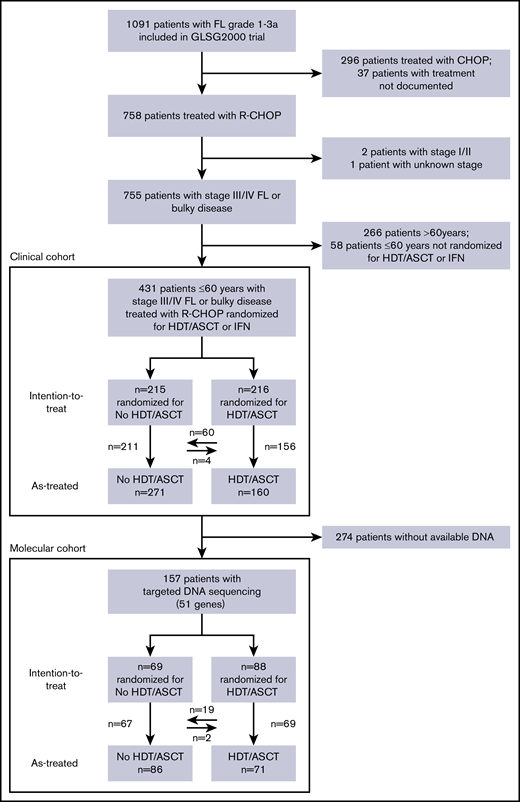
Within the German Low Grade Lymphoma Study Group (GLSG) 2000 trial, medically fit patients ≤60 years with advanced FL were randomized to receive either consolidative HDT/ASCT (total body irradiation, 12 Gy; days –6 to –4; high-dose cyclophosphamide, 60 mg/kg body weight IV, days –3 and –2; reinfusion of peripheral blood stem cells on day 0) or interferon maintenance (5 × 106 U subcutaneously 3 times weekly).11-13 Here, we analyzed only treatment outcomes of the 431 patients who uniformly received immunochemotherapy with R-CHOP (rituximab, 375 mg/m2, day 0; cyclophosphamide, 750 mg/m2, day 1; doxorubicin, 50 mg/m2, day 1; vincristine, 1.4 mg/m2, day 1; prednisone, 100 mg/d, days 1-5; every 3 weeks) as induction regimen, and underwent randomization for HDT/ASCT (Figure 1). All patients had grade 1-3a FL with stage III/IV disease or localized disease considered ineligible for definitive radiotherapy, and were in need of therapy. Additional information regarding treatment is provided in the supplemental Methods. Failure-free survival (FFS) and OS were calculated from date of treatment initiation as previously reported.3,10 FFS events were refractory disease (<partial response) at end of induction treatment, progression, relapse, or death from any cause. The trial was approved by the institutional review board, and all participants gave written informed consent, including for molecular analyses. For our analyses, we used a recent update of the clinical database providing long-term follow-up (data cutoff date: 25 April 2019).

We performed targeted DNA sequencing of all 157 patients with available diagnostic FL biopsies. Sequencing libraries were prepared from >150 ng genomic DNA derived from formalin-fixed paraffin-embedded diagnostic lymphoma tumor specimens. After DNA sonication to 250-bp fragments (Covaris), Illumina sequencing libraries were prepared using SPRIworks reagents (Beckman-Coulter). Enrichment for target regions was achieved using a custom SureSelect hybrid capture panel (Agilent). Three different capture panels were used for samples included in this study. Analyses were restricted to the coding regions of 51 genes represented in all 3 panels (supplemental Methods), including all gene mutations required to calculate the previously established clinicogenetic risk models.3,10 Libraries were sequenced on an Illumina HiSeq2500 machine in Rapid Run mode. We excluded cases with <60% target bases covered ≥30×, and also excluded cases with 60% to 80% target bases covered ≥30× if >0.6% target bases were not covered at all. Data analysis was done using standard data processing tools (Picard tools, Burrows-Wheeler Aligner,14 and Genome Analysis Toolkit.15,16 Single nucleotide variants and short insertions/deletions (indels) were called with MuTect v1.1.4 and SomaticIndelDetector (Genome Analysis Toolkit), respectively. A panel-of-normals filter was generated from normals and nontumor controls, as previously described.10 Variants represented in the panel-of-normals were rejected as putative germline variants or artifacts. Germline polymorphisms from the Exome Sequencing Project and dbSNP databases (build 142) were excluded. We called nonsynonymous variants (missense, frameshift, nonsense, start-codon, and splice site mutations as well as in-frame insertions and deletions) with a variant allele frequency of ≥10%. Mean target coverage for the specimens included in this study was 254X. Median number of genes with ≥1 nonsynonymous mutation per case was 4 (range, 2-15). m7-FLIPI and POD24-PI were calculated from clinical and molecular data, as previously described.3,10
We performed ITT and AT survival and regression analyses to determine the impact of HDT/ASCT on treatment outcome in patients who were risk-stratified by FLIPI, m7-FLIPI, and POD24-PI. We used the binary FLIPI (high risk vs low/intermediate risk) because no significant differences were observed for FFS between low-risk and intermediate-risk patients treated with R-CHOP in previous studies.17,18 The Kaplan-Meier method was used to describe time-to-event endpoints. Cox proportional hazards regression with log-likelihood testing, log-rank test, and logistic regression were applied as indicated.
We used Cox regression considering the interaction term between gene mutation status and treatment (AT) alongside the 2 variables to assess survival benefit mediated by HDT/ASCT for individual genes. To build a multivariable model without prior knowledge of interactions between individual gene mutations we used the random survival forest (RSF)19,20 method to predict FFS in the context of the administered treatment. We used the following features: (1) genes mutated in ≥10% of cases (n = 11), (2) their interaction term with treatment, and (3) treatment administered (AT). The RSF consisted of 1500 trees and, importantly, was implemented in a leave-one-out cross-validation framework to avoid overfitting. For each patient held out from model training, we used the RSF model to estimate the FFS benefit gained from HDT/ASCT based on the patient’s genotype. The benefit score was defined as the expected FFS surplus within the first 5 years when HDT/ASCT was administered (Prediction of Response to Dose-intensified Immunochemotherapy for Follicular Lymphoma [PReDiCt-FL]). Further details are provided in the supplemental Methods section. The benefit score was binarized at its median (PReDiCt-FL low vs PReDiCt-FL high). The RSF variable importance measure was used to quantify the importance of individual features. Mutation frequencies between the 2 predictive groups were compared by Fisher's exact test and adjusted for multiple hypothesis testing by Holm’s method. To validate our methodological approach, we used a previously published method to identify predictive factors based on a proportional hazard approach with a Lasso shrinkage penalty over the variables and interaction terms.21
Results
To explore if we can identify patient subsets with a survival benefit from frontline consolidative HDT/ASCT, we analyzed patients from the GLSG 2000 trial who received rituximab-containing therapy for symptomatic, previously untreated FL, grade 1-3a FL, stage III/IV disease, or localized disease considered ineligible for definitive radiotherapy.
The clinical cohort
A total of 431 patients ≤60 years were randomized to receive consolidative HDT/ASCT (216 patients, 50%) or interferon maintenance (215, 50%) after R-CHOP induction (Figure 1). The cohorts were balanced when considering the distribution of age, sex, FLIPI score, Eastern Cooperative Oncology Group performance status (ECOG PS), and grade (Table 1). Among the 216 patients randomized for HDT/ASCT, 156 (72%) received the treatment as indicated by the study protocol. Median follow-up for OS was 11.2 years, with 95% confidence interval (CI) (10.0-11.7).
HDT/ASCT after R-CHOP does not improve OS in unselected patients
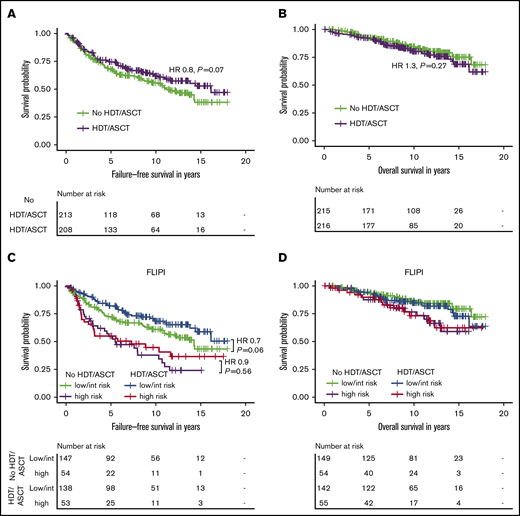
In the intention-to-treat (ITT) analysis, 10-year FFS rates in the HDT/ASCT and no-HDT/ASCT cohorts were 62% (95% CI, 55-70) and 56% (95% CI, 49-64); 10-year OS rates were 80% (95% CI, 74-87) and 85% (95% CI, 79-90), respectively. Patients randomized for consolidative HDT/ASCT had a trend toward longer FFS (hazard ratio [HR], 0.8, P = .07), but OS was not significantly different between the 2 cohorts (HR, 1.3; P= 0.27; Figure 2A-B). When we stratified patients by their actual treatment (AT analysis), the benefit of HDT/ASCT for FFS reached statistical significance (HR, 0.7; P = .04). Of note, even in the AT analysis, we did not even observe a trend toward improved OS with HDT/ASCT. Instead, the trend toward shorter OS was even more pronounced (HR, 1.4; P = .13; supplemental Figure 1).
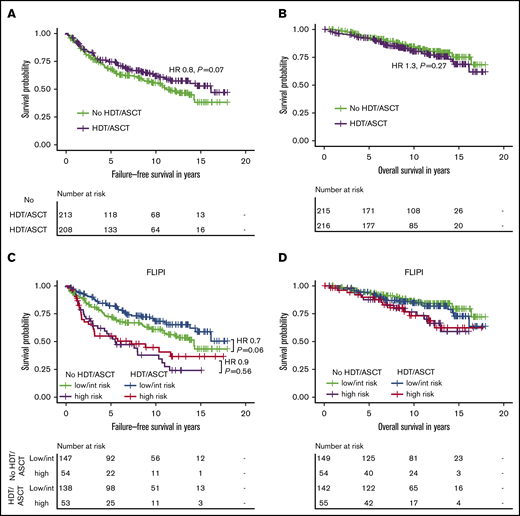
Intention-to-treat analysis for failure-free and overall survival by treatment and FLIPI risk group in the clinical study cohort. Failure-free survival (A) and overall survival (B) by randomization status in 431 patients receiving R-CHOP within the GLSG2000 trial. Failure-free survival (C) and overall survival (D) by randomization status and FLIPI risk group. The hazard ratio and P value given in panel C are derived from Cox regression.
Intention-to-treat analysis for failure-free and overall survival by treatment and FLIPI risk group in the clinical study cohort. Failure-free survival (A) and overall survival (B) by randomization status in 431 patients receiving R-CHOP within the GLSG2000 trial. Failure-free survival (C) and overall survival (D) by randomization status and FLIPI risk group. The hazard ratio and P value given in panel C are derived from Cox regression.
HDT/ASCT after R-CHOP does not improve OS in patients with high-risk FLIPI
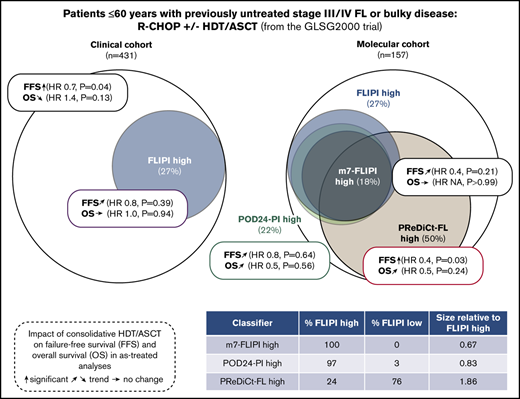
Because no survival difference was observed in unselected patients, we sought to analyze whether a high-risk cohort may benefit from HDT/ASCT. Only 109 of 400 evaluable patients (27%) were classified as high risk according to the FLIPI. Although patients with FLIPI low/intermediate risk had a trend toward longer FFS with HDT/ASCT (HR, 0.7; P = .06), no difference was observed in the high-risk cases (HR, 0.9; P = .56). Again, there was no significant difference for OS with HDT/ASCT (low/intermediate risk: HR, 1.3; P = .41; high-risk: HR, 1.0; P = 1.0; Figure 2C-D). Furthermore, the frequencies of POD24 did not significantly differ between treatment arms (12% vs 16%, P = .39; Table 1). Similar results were obtained in the corresponding AT analysis (supplemental Table 1; supplemental Figure 1), which also failed to demonstrate a significant benefit associated with HDT/ASCT.
Molecular cohort
We then analyzed whether integrating somatic gene mutations and clinical factors may be capable of identifying patients benefiting from HDT/ASCT. A total of 157 of the 431 patients were available for molecular analyses. Among this genotyped subset, 88 patients (56%) were randomized to receive consolidative HDT/ASCT, whereas 69 (44%) were randomized to interferon maintenance. Of the 88 patients randomized for HDT/ASCT, 69 (78%) eventually received treatment as indicated by the study protocol, whereas 22% opted against such therapy. Among the former, 2 of the 69 patients (3%) randomized for interferon maintenance treatment also proceeded with HDT/ASCT and crossed over (Figure 1).
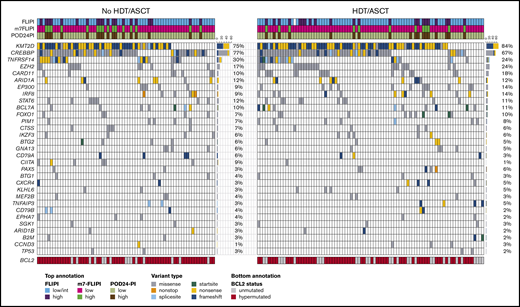
In the ITT analysis, the HDT/ASCT and no-HDT/ASCT cohorts were balanced when considering distribution in age (51 vs 50 years), sex (50% vs 64% male), high-risk FLIPI (25% vs 29%), and ECOG PS >1 (6% vs 6%; Table 1). Recurrent gene mutation frequencies were also similar between the 2 cohorts, including mutations in prognostically relevant genes such as TP53 (2% vs 3%) and EZH2 (24% vs 17%) (Table 1; Figure 3). The most frequently mutated other genes in the 2 cohorts were KMT2D (84% vs 75%), CREBBP (67% vs 77%), TNFRSF14 (24% vs 30%), CARD11 (18% vs 10%), ARID1A (12% vs 12%), and EP300 (14% vs 9%) (Figure 3). Patient characteristics for the AT cohorts are summarized in supplemental Table 1. The outcome of the molecular cohort was largely representative of the clinical cohort. For example, 10-year FFS rates in the molecular ITT HDT/ASCT and no-HDT/ASCT cohorts were 62% and 53% (HR, 0.8; P = .28), whereas 10-year OS rates were 87% and 81% (HR, 0.9; P = .71; supplemental Figure 2), respectively. When compared with these ITT analyses, similar patterns were also evident in AT analysis (supplemental Figure 3).
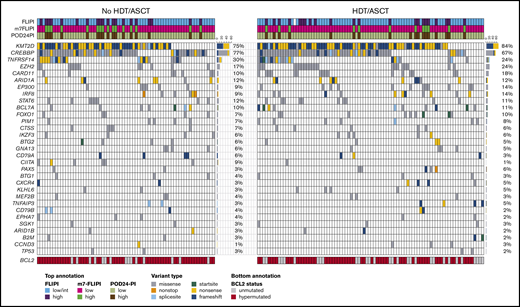
Targeted mutational landscape of the molecular study cohort. The Oncoprint shows recurrently mutated genes as rows and the 157 patients of the molecular study cohort as columns by randomization status. The color coding delineates the type of nonsynonymous alteration detected by targeted next-generation sequencing of 51 genes. The top annotation indicates FLIPI, m7-FLIPI, and POD24-PI risk groups. For BCL2 (bottom annotation), all variants including silent and noncoding variants were reported as hypermutations. The number of cases with respective gene mutations and corresponding mutation frequencies by cohort are visualized on the right side of the Oncoprints.
Targeted mutational landscape of the molecular study cohort. The Oncoprint shows recurrently mutated genes as rows and the 157 patients of the molecular study cohort as columns by randomization status. The color coding delineates the type of nonsynonymous alteration detected by targeted next-generation sequencing of 51 genes. The top annotation indicates FLIPI, m7-FLIPI, and POD24-PI risk groups. For BCL2 (bottom annotation), all variants including silent and noncoding variants were reported as hypermutations. The number of cases with respective gene mutations and corresponding mutation frequencies by cohort are visualized on the right side of the Oncoprints.
Low rates of patients reclassified by m7-FLIPI and POD24-PI in HDT/ASCT-eligible cohort
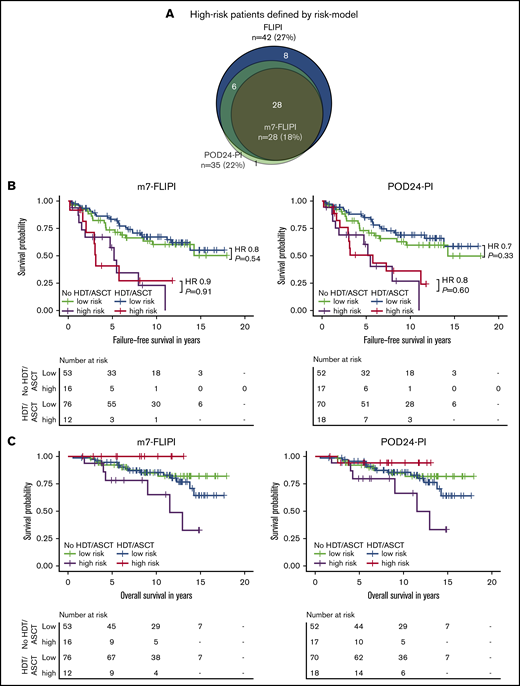
We then evaluated the clinicogenetic risk models m7-FLIPI and POD24-PI in young, transplant-eligible patients. Patients classified as being high risk as identified by FLIPI, m7-FLIPI, and POD24-PI comprised only 27% (n = 42), 18% (n = 28), and 22% (n = 35) of the cohort, respectively (Figure 4A). The m7-FLIPI reclassified only 9% (n = 14) of patients from high-risk FLIPI to low-risk m7-FLIPI. The POD24-PI reclassified only 5% (n = 8) of patients from high-risk FLIPI to low-risk POD24-PI; 1 patient was reclassified from low-/intermediate-risk FLIPI to high-risk POD24-PI.
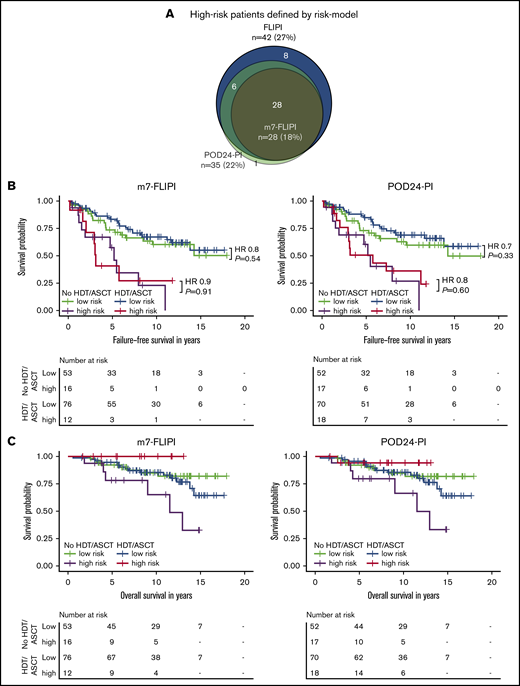
Intention-to-treat analysis for failure-free and overall survival by risk group assessed by m7-FLIPI and POD24-PI and treatment. (A) Scaled Venn diagram depicting sizes and relations of high-risk cohorts identified by FLIPI, m7-FLIPI, and POD24-PI in 156 patients evaluable for all 3 scores. Failure-free survival (B) and overall survival (C) by m7-FLIPI and POD24-PI risk group and randomization status. Hazard ratios and P values given in the graph are derived from Cox regression.
Intention-to-treat analysis for failure-free and overall survival by risk group assessed by m7-FLIPI and POD24-PI and treatment. (A) Scaled Venn diagram depicting sizes and relations of high-risk cohorts identified by FLIPI, m7-FLIPI, and POD24-PI in 156 patients evaluable for all 3 scores. Failure-free survival (B) and overall survival (C) by m7-FLIPI and POD24-PI risk group and randomization status. Hazard ratios and P values given in the graph are derived from Cox regression.
m7-FLIPI and POD24-PI stratify FFS in young, transplant-eligible FL patients
The risk to develop POD24 was increased in patients classified as high risk by any of the 3 indices (FLIPI: odds ratio, 5.3, P = .003; m7-FLIPI: 5.5, P = .003; POD24-PI: 5.1, P = .004). Accordingly, high-risk patients experienced shorter FFS (FLIPI: HR, 2.9, P < .001; m7-FLIPI: HR, 3.5, P < .001; POD24-PI: HR, 3.1, P < .001; supplemental Figure 4A). OS was not significantly different when considering high-risk status using these 3 indices against other risk strata (FLIPI: HR, 1.6, P = .25; m7-FLIPI: HR, 1.7, P = .25; POD24-PI: HR, 1.6, P = .30; supplemental Figure 4B).
m7-FLIPI and POD24-PI are not optimized to identify high-risk patients who benefit from HDT/ASCT
Importantly, consolidative HDT/ASCT did not significantly prolong FFS in high-risk patients as defined by m7-FLIPI (HR 0.9, P = .91) and POD24-PI (HR 0.8, P = .60; Figure 4B). Similarly, OS was not significantly improved in the high-risk subsets (m7-FLIPI: HR, NA, P = .05; POD24-PI: HR, 0.2, P = .11; Figure 4C). Equally, HDT/ASCT was neither associated with significantly prolonged FFS In low-risk patients (m7-FLIPI: HR, 0.8, P = .54; POD24-PI: HR, 0.7, P = .33; Figure 4B), nor improved OS (m7-FLIPI: HR, 1.4, P = .48; POD24-PI: HR, 1.3, P = .50; Figure 4C). Supplemental Figure 5 summarizes the corresponding AT analyses.
PReDiCt-FL: a molecular model to predict response to dose-intensified immunochemotherapy
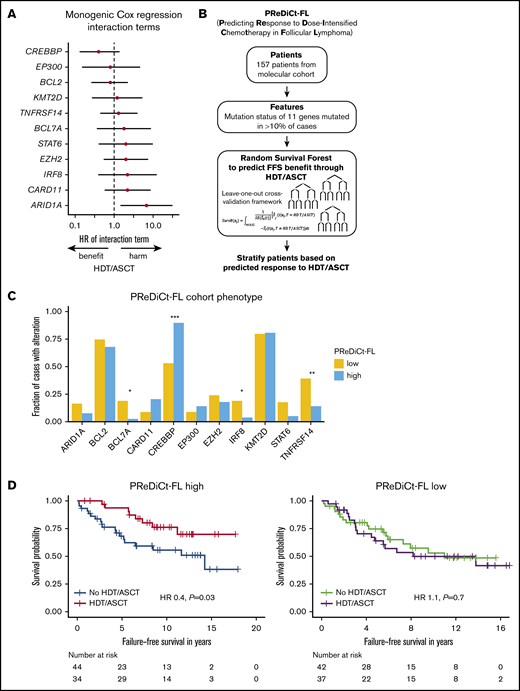
We next explored whether distinct genotypes could predict treatment outcome after HDT/ASCT. No individual gene significantly predicted FFS after HDT/ASCT when adjusting for multiple hypothesis correction, but there was a trend toward longer FFS through HDT/ASCT in CREBBP-mutated cases. An opposite trend was observed for ARID1A mutations (Figure 5A). As an exploratory exercise to predict therapy-specific FFS, we built multivariable models without a priori knowledge of possible interactions between gene mutations in the context of therapy. Specifically, we used RSF to test for such interactions. Importantly, we used a leave-one-out cross-validation framework to avoid overfitting. Considering the mutation status of each of the 11 genes with mutations in ≥10% of cases, we estimated the expected FFS benefit from HDT/ASCT for each patient in a novel model (PReDiCt-FL; Figure 5B). CREBBP mutation status had the highest variable importance among all features included in this model (supplemental Figure 6A), and was also identified in a proportional hazard approach with Lasso shrinkage (supplemental Table 2). We stratified patients based on their expected FFS advantage by HDT/ASCT. Interestingly, cases with high PReDiCt-FL scores (ie, greater than median predicted benefit) were significantly enriched for CREBBP mutations (P = 4.3 × 10−6) and depleted for TNFRSF14 (P = .005), BCL7A (P = .01), and IRF8 (P = .04) mutations (Figure 5C). Patients with high PReDiCt-FL scores had longer FFS with HDT/ASCT (HR, 0.4; P = .03). In contrast, cases with low PReDiCt-FL scores (ie, lower than the median) had no FFS benefit from HDT/ASCT (HR, 1.1; P = .7; Figure 5D). Of note, 58 of 78 patients with high PReDiCt-FL scores (74%) were not classified as high-risk by FLIPI, m7-FLIPI, and POD24-PI (supplemental Figure 6C). Importantly, no significant differences were observed for OS (PReDiCt-FL high: HR, 0.5; P = .24; supplemental Figure 6B).

Prediction of failure-free survival in the context of HDT and ASCT. (A) Forest plot depicting HRs and 95% CIs of interaction terms of monogenic Cox regressions for failure-free survival. Rows represent results of individual Cox regressions considering treatment (as-treated), mutations status of the respective gene, and the interaction term. HRs and 95% CIs of the interaction terms are visualized. Only genes mutated in ≥10% of cases (n = 11) were included in the analysis. (B) Development of a multivariable model to predict failure-free survival benefit from HDT/ASCT (PReDiCt-FL). (C) Comparison of mutation frequencies between PReDiCt-FL low (yellow) and PReDiCt-FL high (blue) cohorts. Asterisks indicate significance levels assessed by Fisher’s exact test with Holm correction. (D) FFS of cases predicted to have a high FFS benefit (PReDiCt-FL high, left) and predicted to have no benefit from HDT/ASCT (PReDiCt-FL low, right), respectively. *P < .05; **P < .01; ***P < .001.
Prediction of failure-free survival in the context of HDT and ASCT. (A) Forest plot depicting HRs and 95% CIs of interaction terms of monogenic Cox regressions for failure-free survival. Rows represent results of individual Cox regressions considering treatment (as-treated), mutations status of the respective gene, and the interaction term. HRs and 95% CIs of the interaction terms are visualized. Only genes mutated in ≥10% of cases (n = 11) were included in the analysis. (B) Development of a multivariable model to predict failure-free survival benefit from HDT/ASCT (PReDiCt-FL). (C) Comparison of mutation frequencies between PReDiCt-FL low (yellow) and PReDiCt-FL high (blue) cohorts. Asterisks indicate significance levels assessed by Fisher’s exact test with Holm correction. (D) FFS of cases predicted to have a high FFS benefit (PReDiCt-FL high, left) and predicted to have no benefit from HDT/ASCT (PReDiCt-FL low, right), respectively. *P < .05; **P < .01; ***P < .001.
Discussion
Current guidelines do not recommend consolidative HDT/ASCT for patients with advanced FL in first remission after state-of-the-art immunochemotherapy.8,9 So far, however, remarkably few data were available to support this recommendation in the era of modern immunochemotherapy, in particular with respect to high-risk cohorts. Here, we provide definitive evidence against frontline HDT/ASCT in unselected patients, since ITT and AT analyses did not show an OS benefit in our large randomized trial with mature follow-up. Our study also did not demonstrate an OS benefit for high-risk cohorts as defined by the clinicogenetic risk models m7-FLIPI and POD24-PI. Finally, we describe a novel machine-learning model to predict benefit from HDT/ASCT based on the mutation status of 11 genes (PReDiCt-FL), which identifies patients with improved FFS, but, again, not OS.
The efficacy of consolidative HDT/ASCT for patients with previously untreated advanced FL has been investigated in 4 randomized clinical studies,22-25 but only 1 trial has been conducted since the addition of anti-CD20 antibodies has become standard of care. The Italian GITMO/IIL trial reported on 134 evaluable patients and demonstrated longer PFS for a rituximab-HDT/ASCT approach compared with 6 cycles of CHOP-rituximab, but OS was not significantly different.25,26 In our larger study including 431 patients, we also observed a small improvement of FFS, which only reached statistical significance in the AT analysis. However, because of the significant treatment toxicity associated with HDT/ASCT, OS should be the only acceptable end point to change current practice. After a follow-up of >10 years, the modest benefit seen for FFS with HDT/ASCT did not translate into prolonged OS. In fact, we observed a trend toward shorter OS in patients randomized for HDT/ASCT, which was even more pronounced in the AT analysis. This seems particularly remarkable because AT cohorts for HDT/ASCT can be expected being enriched for a more favorable patient collective. Collectively, our mature data provide the most definitive evidence supporting current guidelines, that HDT/ASCT should not be offered to unselected patient cohorts in the frontline setting.
In contrast, HDT/ASCT is an effective salvage treatment of early relapsed FL after initial immunochemotherapy and recommended for patients with chemosensitive disease who are considered eligible for dose-intensified approaches by most current treatment guidelines.5-9 Because the clinicogenetic risk scores m7-FLIPI and POD24-PI have been shown to enrich for patients with early treatment failure, we reasoned that high-risk patients may benefit from frontline HDT/ASCT. To the best of our knowledge, this is the first study to address this hypothesis. Despite the young age of our patient cohort, the mutational landscape described here is remarkably similar to other unselected cohorts.10,27-32 This is in line with our previously reported finding that patient age at diagnosis does not significantly affect disease biology in FL.33 In this study, we could not demonstrate a FFS or OS benefit from frontline HDT/ASCT for high-risk patients as defined by FLIPI, m7-FLIPI and POD24-PI, both by ITT and AT analyses.
The m7-FLIPI and POD24-PI integrate the FLIPI score and gene mutation status to stratify for FFS. In unselected cohorts of patients with advanced FL, both scores have been shown to outperform the FLIPI by reclassifying patients into low-risk groups. In prior studies, the fraction of high-risk FLIPI was remarkably stable at ∼50%, with about one-half of these being reclassified to low risk by the m7-FLIPI.10,34 In contrast, this study in young and medically fit patients shows that the fraction of patients identified to be high risk by FLIPI is low (<30%). Furthermore, the fractions of patients being reclassified by the m7-FLIPI and POD24-PI was low (<10%). The underlying reason for this observation is that the performance of the FLIPI is age-dependent,18 and so are the m7-FLIPI and POD24-PI. Our data demonstrate that these established clinicogenetic-risk models are not suited to stratify younger patient cohorts.
To investigate whether genotypes may be predictive of outcome after HDT/ASCT, we built a machine-learning model to predict FFS in the context of intensified frontline treatment (PReDiCt-FL). Using cross-validation to avoid overfitting, PReDiCt-FL identified a subset of patients benefiting from HDT/ASCT. However, even the observed FFS benefit from HDT/ASCT in patients with higher PReDiCt-FL scores did not translate into a statistically significant and clinically relevant impact on OS. Of note, more than one-half of the patients predicted to have an FFS benefit from HDT/ASCT were in fact not identified to be high risk by FLIPI, m7-FLIPI, and POD24-PI. This indicates that the determinants of FFS benefit from frontline HDT/ASCT are distinct from the previously identified features associated with higher risk of early treatment failure.
CREBBP mutation status had the highest importance in the PReDiCt-FL model, and cases predicted to have improved FFS with HDT/ASCT were highly enriched for CREBBP mutations and depleted for TNFRSF14 mutations. Interestingly, both mutations have previously been shown to affect the crosstalk of FL cells with its immune microenvironment. CREBBP mutations downregulate MHC II expression and, among others, lead to immune evasion in B-cell lymphomas.31,35-37 TNFRSF14 mutations, on the other hand, are involved in recruiting T-follicular helper cells creating a tumor supportive microenvironment.38 Interestingly, high-dose cyclophosphamide, which was part of the HDT administered in this study, has been shown to induce an acute secretory activating phenotype resulting in macrophage infiltration and increased sensitivity of tumors to treatment with monoclonal antibodies.39,40 It is therefore tempting to speculate that patients with high PReDiCt-FL scores might benefit from HDT/ASCT because of the modulating capacity of high-dose cyclophosphamide on the FL tumor microenvironment. If so, PReDiCt-FL may be a useful tool to more broadly identify patient subsets who may benefit from distinct acute secretory activating phenotype-inducing therapies other than HDT/ASCT, potentially also in the relapsed/refractory setting and in other lymphomas. Moreover, it may be highly informative to better understand the cellular and molecular mechanisms of how individual components of HDT (eg, high-dose cyclophosphamide) affect the interplay between lymphoma cells and components of the immune microenvironment and ultimately treatment outcome in molecularly defined FL subtypes, to develop non-HDT/ASCT approaches with similar efficacy but less toxicity. This may also apply to emerging immune modulating therapies and CAR-T cells.41-44
Our data suggest that molecularly defined subtypes of FL may be more susceptible to dose-intensified frontline treatment as relates to disease control and progression. However, we did not observe a statistically significant difference in OS, which may have several reasons. In particular, successful salvage strategies including HDT/ASCT for relapsed/refractory FL may have affected our results. Moreover, prediction accuracy of genotype-based pretreatment risk models may be insufficient to identify patient subsets with OS benefit from HDT/ASCT. Positron emission tomography (PET) scans at baseline and during/after treatment are strong prognostic factors in FL,45-48 but were not obtained in this trial, which was initiated more than 20 years ago.11 Future studies in FL should combine pre- and on-treatment biomarkers (eg, molecular risk scores, baseline and follow-up PET scans, minimal residual disease assessment49 ) into dynamic risk models to identify patients at highest risk for treatment failure before overt relapse, to evaluate intensified or innovative treatment approaches, as recently proposed in other B-cell malignancies.50
Although we cross-validated our model to avoid overfitting of PReDiCt-FL, our results are hypothesis-generating and require independent validation. Nevertheless, our methodological approach to identifying a predictive biomarker could serve as a blueprint for future studies that aim to establish predictive biomarkers for state-of-the-art treatment alternatives (eg, different anti-CD20 antibodies [rituximab vs obinutuzumab], chemotherapy-backbones [CHOP vs bendamustine]) in FL.
We realize that the size of our molecular cohort, and particularly the small subset of patients classified as high risk certainly is a limitation of our study, affecting the power to detect differences in treatment outcomes mediated by HDT/ASCT. However, because of the paucity of cohorts with available biobanked biopsy material in this setting, this study will likely be the largest ever to address this question. Therefore, these data will be highly valuable as a benchmark for future clinical trials.
In conclusion, our data from a large randomized trial with long follow-up provide the most definitive evidence that consolidative HDT/ASCT should not be offered to unselected patients in first remission after R-CHOP. Furthermore, we could not identify patient subsets with OS benefit from frontline HDT/ASCT by clinical and genotype-based risk models. Further research is warranted to evaluate the use of molecular profile-guided therapeutic approaches for FL.
Patient and sequencing data that support the findings of this study are available upon request from the corresponding author, Oliver Weigert, at oliver.weigert@med.uni-muenchen.de.
Acknowledgments
S.A. is funded by a Dr. Mildred Scheel postdoctoral fellowship of the Deutsche Krebshilfe (57406718). O.W. is supported by the Max-Eder Program of the Deutsche Krebshilfe (70110659/70112997).
Authorship
Contribution: S.A. and O.W. designed and performed the research, analyzed data, and wrote the manuscript; V.J., M.S.E., and E.H. oversaw statistical analyses; N.T., A.Z., C.S., and M.U. were involved in collecting and analyzing clinical data; S.H., V.P., J.C.H., E.G., and W. Keay performed analyses; A.R., W. Klapper, H.S., A.F., G.O., A.M.S., H.H., M.L.H., and C.P. provided samples; and M.D., A.A.A., and W.H. supervised the study and contributed to the preparation of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Oliver Weigert, Department of Medicine III, LMU Hospital, Laboratory for Experimental Leukemia and Lymphoma Research (ELLF), Max-Lebsche Platz 30, 81377 Munich, Germany; e-mail: oliver.weigert@med.uni-muenchen.de.