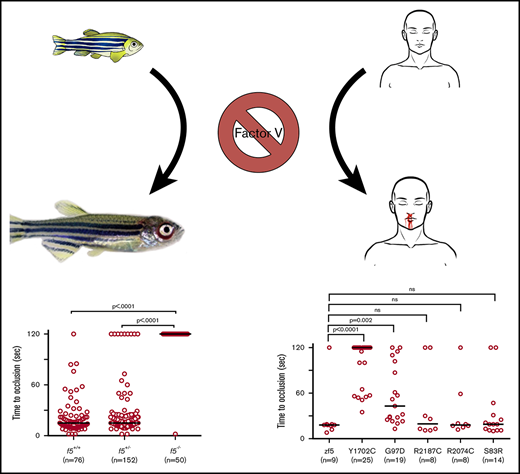
Key Points
f5 mutant fish embryos tolerate symptoms lethal in mammals but succumb to bleeding in adulthood.
Analysis of patient F5 variants demonstrate that all have some residual ability to restore hemostasis in response to endothelial injury.

Abstract
In humans, coagulation factor V (FV) deficiency is a rare, clinically heterogeneous bleeding disorder, suggesting that genetic modifiers may contribute to disease expressivity. Zebrafish possess many distinct advantages including high fecundity, optical clarity, external development, and homology with the mammalian hemostatic system, features that make it ideal for genetic studies. Our aim was to study the role of FV in zebrafish through targeted mutagenesis and apply the model to the study of human F5 variants. CRISPR-mediated genome editing of the zebrafish f5 locus was performed, generating mutants homozygous for a 49 base pair deletion in exon 4. Thrombus formation secondary to vascular endothelial injury was absent in f5−/− mutant embryos and larvae. Despite this severe hemostatic defect, homozygous mutants survived before succumbing to severe hemorrhage in adulthood. Human F5 variants of uncertain significance from patients with FV deficiency were evaluated, and the causative mutations identified and stratified by their ability to restore thrombus formation in larvae. Analysis of these novel mutations demonstrates variable residual FV function, with minimal activity being required to restore hemostasis in response to laser-induced endothelial injury. This in vivo evaluation may be beneficial for patients whose factor activity levels lack correlation with bleeding symptomatology, although limitations exist. Furthermore, homozygous mutant embryos tolerate what is a severe and lethal defect in mammals, suggesting the possibility of species-specific factors enabling survival, and allowing further study not possible in the mouse. Identification of these factors or other genetic modifiers could lead to novel therapeutic modalities.
Introduction
Coagulation factor V (FV, gene F5) is an essential component of hemostasis. In plasma, it circulates as a large 330 kDa glycoprotein that shares homology and a similar domain organization with factor VIII.1,2 The inactive pro-cofactor form of FV is synthesized in the liver as a 2224 amino acid polypeptide, secreted as a 2196 amino acid single chain protein, and the large B domain is removed during activation. In its activated form, FVa serves as the cofactor protein for the activated serine protease factor Xa (FXa, gene F10) in the prothrombinase complex, which rapidly converts prothrombin to thrombin. The gene for FV is located on the long arm of chromosome 1, band 23, consists of 25 exons, and is ∼80 kb in length.3
Congenital deficiency of FV in humans is a rare autosomal recessive bleeding disorder that affects 1 in 1 000 000 individuals, the vast majority resulting from null mutations.4-7 FV deficiency is divided into type I (quantitative) and type II (qualitative) defects, with the majority of mutations falling in the former group.8 Treatment options are limited, because only recently has a purified FV concentration been developed.9 Multifactor concentrates contain inadequate concentrations of FV to be effective. For major bleeding, fresh frozen plasma or platelet transfusions are typically used.4 For minor bleeding, antifibrinolytics are often sufficient.6
Insight into the crucial role of FV has been facilitated using gene targeting in mice. F5 knockouts exhibit complete lethality, with death in either the embryonic or perinatal periods,10 although transgenic expression of very small amounts (<0.1%) of F5 rescues this phenotype.11 In humans, levels of FV antigen do not correlate well with bleeding symptomatology. In patients with severe FV deficiency (levels <1%), mucosal bleeding is seen most commonly. Fewer patients present with menorrhagia, hematomas, and hemarthroses.12 The relatively mild bleeding seen in many patients with severe deficiency may be explained by findings that <1% FV is adequate to ensure minimal thrombin generation as demonstrated in in vivo,13 in vitro,14 and in silico15 studies. However, this does not explain the striking variability in phenotype between patients with equivalent FV levels. This variability may be explained in some patients by residual platelet FV, as well as low levels of tissue factor pathway inhibitor.16,17
Zebrafish (Danio rerio) have been used extensively to study hemostasis and have demonstrated significant homology with the mammalian coagulation system. They possess distinct advantages, including high fecundity, optical clarity, and external development. New developments in genome editing technology have facilitated quick and robust systems for targeted genetic modification.18-20 We and others have demonstrated the benefits of this system for modeling hemostasis and thrombosis.21-33 Knockout models have demonstrated longer than expected survival in comparison with their mammalian counterparts,24,34,35 enabling additional characterization of the phenotypes. We previously demonstrated a severe hemostatic defect in FX-deficient zebrafish early in development with lack of venous occlusion in response to endothelial injury at 3 days postfertilization (dpf).35 Homozygous mutants tolerate this defect with no overt hemorrhage until 3 to 4 weeks of age and survival for several months. Anticoagulant deficiency has also been shown to exhibit milder phenotypes in the zebrafish in comparison with murine models. A zebrafish model of antithrombin (At3) deficiency exhibited a phenotype similar to disseminated intravascular coagulation.24 Despite this, homozygous mutants lived to adulthood, in contrast to the prenatal mortality observed in mice.24,36 Here we report targeted ablation of zebrafish f5 using the CRISPR/Cas9 genome editing platform. Homozygous mutant embryos and larvae demonstrate a severe defect in hemostasis. They tolerate this into adulthood, but eventually succumb to lethal bleeding by 6 months of age.
Methods
Zebrafish strains and maintenance
Zebrafish were raised in accordance with animal care guidelines as approved by the University of Michigan Animal Care and Use Committee. Embryos are defined as 0 to 2 dpf, larvae 3 to 29 dpf, juvenile 30 to 89 dpf, and adult ≥90 dpf.37 f5 mutant zebrafish were generated on an AB × TL hybrid background.
Targeted mutagenesis of the f5 locus using CRISPR/Cas9 genome editing
The f5 locus was identified in the zebrafish genomic sequence assembly38 and CRISPR/Cas9-mediated genome editing was used to induce mutations in exon 4. Target sites of 17 to 20 nucleotides were identified using the Web-based ZiFiT Targeter program (http://zifit.partners.org) as described elsewhere.39 Templates for synthetic single-guide RNAs (sgRNAs) were constructed using polymerase chain reaction (PCR) to fuse overlapping oligonucleotides (Integrated DNA Technologies, Coralville, IA), which contained the complementary 20 nucleotides of the target site, the T7 promoter, and linearized vector pDR27440 as a template (Table 1). The resulting double-stranded DNA fragment (120 base pairs) was then column purified and transcribed using the T7 Quick High Yield RNA Synthesis kit (New England BioLabs, Ipswich, MA) and purified using RNA Clean and Concentrator kit (Zymo Research, Irvine, CA). Cas9 messenger RNA (mRNA) was transcribed as previously described.20 sgRNAs and Cas9 mRNA were injected concurrently into the cytoplasm of 1-cell stage embryos at concentrations of 12.5 and 300 ng/µL, respectively. The resulting F0 population was raised to adulthood and crossed with wild-type zebrafish to verify germline transmission to the F1 generation.
Genotyping of f5 mutant offspring
Staged zebrafish larvae or adults were anesthetized in tricaine (0.16 mg/mL, Western Chemical Inc., Ferndale, WA) or humanely euthanized in high-dose tricaine (1.6 mg/mL). Fish were tail fin-clipped as described previously.24,41 Genomic DNA was isolated by incubating tissue in lysis buffer (10 mM Tris-Cl, pH 8.0; 2 mM EDTA, 2% Triton X-100, and 100 µg/mL proteinase K) at 55°C overnight, followed by proteinase K inactivation at 95°C for 5 minutes.24 Insertion and deletion mutations were detected by PCR and resolved by agarose gel electrophoresis or using a Qiaxcel (Qiagen, Hilden, Germany). Primers were designed using Primer342 and oligonucleotides ordered from Integrated DNA Technologies (Table 1).
Laser-induced endothelial injury in zebrafish larvae
Laser-mediated endothelial injury was performed on the posterior cardinal vein (PCV) of zebrafish larvae at 3 dpf as described.25 Larvae were anesthetized in tricaine, embedded in 0.8% low-melt agarose on glass cover slips, and the endothelium of the PCV at the fifth somite distal to the anal pore was ablated using 100 pulses at power level 18 (MicroPoint Pulsed Laser System, Andor Technology, Belfast, Northern Ireland). Arterial injury was also performed on the dorsal aorta at the fifth somite distal to the anal pore at 9 dpf and endothelial injury to the primordial midbrain channel (PMBC) just lateral to the otic capsule at 6 to 7 dpf. The time to occlusion was recorded for up to 2 minutes by a blinded observer. Larvae were subsequently recovered from agarose and genotyped.
Construction of f5 in vivo expression vectors
pDestTol2pA2_ubi:p2A-EGFP35 contains a BbsI restriction site flanked multiple cloning site and a self-cleaving p2A peptide 5′ to the egfp sequence. The vector was linearized using BbsI and purified. The D rerio f5 complementary DNA (cDNA) was amplified from plasmid MDR1734-202728340 (Dharmacon, Lafayette, CO) using primers with f5 and vector homology (Table 1). The resulting 6.3-kb product was purified and inserted into BbsI linearized pDestTol2pA2_ubi:p2A-EGFP using In-Fusion Cloning (Clontech, Mountainview, CA) and transformed into high efficiency 10-β competent Escherichia coli (New England BioLabs) to generate pDestTol2pA2_ubi:f5-p2A-EGFP. Positive colonies were grown in LB media supplemented with 50 µg/mL ampicillin and sequence verified. Mutations were introduced using the QuickChange XL II site-directed mutagenesis kit (Agilent) and mutation-specific primers (Table 1). Amino acid numbering reflects the human amino acid position.
One-cell injection of f5 expression vectors
Circular f5 expression plasmids (10 ng/µL) and transposase mRNA (50 ng/µL)43 were coinjected into 1-cell stage embryos from f5Δ49+/− incrosses. Fluorescent embryos were selected at 3 dpf for laser-mediated endothelial injury.
Whole mount in situ hybridization
Inhibition of pigmentation in embryos and larvae was accomplished using 0.2 mM phenylthiourea (Sigma-Aldrich, St. Louis, MO) at 24 hours postfertilization. Staged larvae were fixed in 4% paraformaldehyde in phosphate-buffered saline with 0.01% Tween-20 at 4°C overnight. They were then dehydrated stepwise into 100% ethanol. Partial f5 cDNA fragments were amplified to synthesize sense and antisense digoxin-labeled RNA riboprobes (DIG RNA labeling kit, Roche, Basel, Switzerland) (Table 1). Amplification of cDNA templates, synthesis of riboprobes, and whole mount in situ hybridization were performed as previously described.24,44,45
RNA isolation, cDNA generation, and reverse transcription PCR
Larvae at 4 dpf were euthanized in high-dose tricaine, tissue removed for genotyping, and then placed in RLT plus buffer (Qiagen); 21 of each genotype were divided into 3 pools of 7 for RNA extraction. Total RNA was isolated using RNEasy Plus Mini Kit (Qiagen) and reverse transcribed with Superscript III Synthesis (Invitrogen). cDNAs were amplified using PrimeTime qPCR Gene Expression Master Mix (Integrated DNA Technologies).
Chemical treatment of embryos
Embryos were incubated in 100 mM ε-aminocaproic acid (Sigma-Aldrich) dissolved in system water starting at 24 hours postfertilization and continuing until 3 dpf. At 3 dpf, they were evaluated by laser-mediated endothelial injury.
Results
Targeted disruption of f5 using genome editing nucleases results in spontaneous hemorrhage and adult lethality
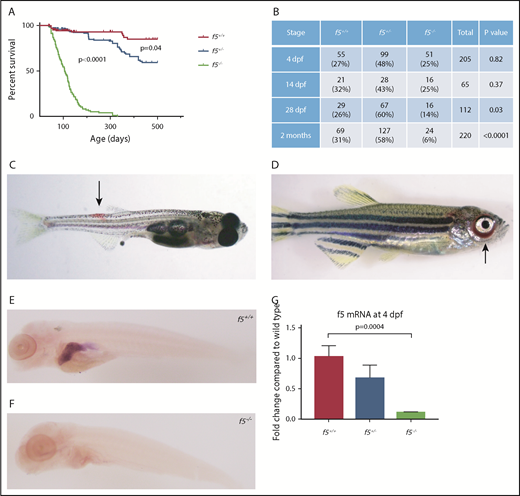
We used CRISPR-Cas9 genome editing to target f5. Several sgRNAs were constructed to target exon 4 of the zebrafish f5 genomic locus. The most robust sgRNA resulted in somatic mutations at a frequency of 66.4%. The resulting embryos were raised to adulthood and F0 founders were mated to wild-type fish to verify germline transmission. We identified many potential founders and verified 1 for further analysis, a 49-bp deletion (f5Δ49 hereafter referred to as f5−) within the predicted A1 domain (Figure 1A-B). The mutation was confirmed both at the genomic and cDNA levels (Figure 1C-D). Heterozygotes from each line were incrossed, and genotyping before 1 month of age revealed a normal Mendelian distribution. Beginning at 28 dpf, we observed a statistically significant loss of homozygotes, and by 7 months postfertilization (mpf), ∼90% died (P < .0001) (Figure 2A-B). Interestingly, we saw a significant loss of heterozygotes starting around 18 months of age (P = .04). Spontaneous hemorrhage was observed grossly in homozygous mutants beginning around 28 dpf, coincident with the beginning of observed lethality (Figure 2C-D).
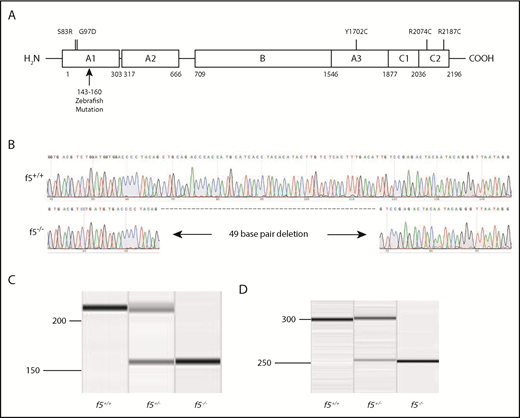
Targeting of the f5 locus using genome editing produces a null allele. (A) Structure of human F5; residue numbering is from the human protein. The CRISPR/Cas9-induced mutation deletes what would be human residues 143 through 160 in the A1 domain and induces a frameshift. Numbered positions indicate location of human mutations evaluated in Figure 5. (B) Sequence of 49-bp f5 exon 4 deletion. (C-D) Amplification of genomic and cDNA from f5+/+, f5+/−, and f5−/− fish demonstrates the 49-bp deletion with no evidence for any other products.
Targeting of the f5 locus using genome editing produces a null allele. (A) Structure of human F5; residue numbering is from the human protein. The CRISPR/Cas9-induced mutation deletes what would be human residues 143 through 160 in the A1 domain and induces a frameshift. Numbered positions indicate location of human mutations evaluated in Figure 5. (B) Sequence of 49-bp f5 exon 4 deletion. (C-D) Amplification of genomic and cDNA from f5+/+, f5+/−, and f5−/− fish demonstrates the 49-bp deletion with no evidence for any other products.
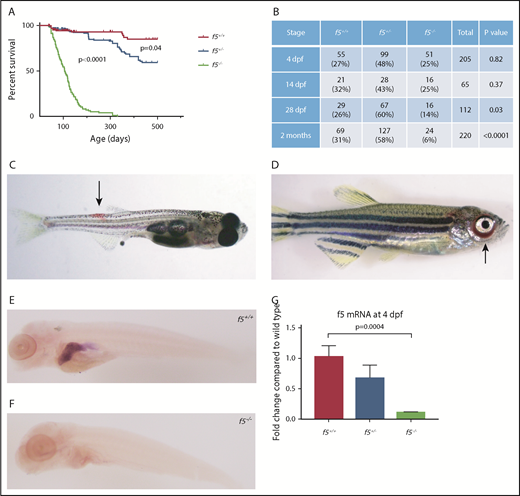
Phenotypes of f5 homozygous mutants reveal adult lethality and spontaneous hemorrhage in homozygous mutant adult fish. (A) Survival curves of zebrafish offspring from f5+/− incrosses, starting at 2 months of age, demonstrate progressive loss of 90% of homozygotes by 7 mpf. Heterozygotes also demonstrate statistically significant decreased survival compared with wild-type fish, with 50% mortality by 1.5 years of age. (B) Genotype distributions of offspring from f5+/− incrosses evaluated at various stages demonstrate loss of homozygotes beginning at 28 dpf. (C) Spontaneous hemorrhage distal to the dorsal fin (arrow) in a 4-week-old homozygous mutant fish. (D) Severe spontaneous periorbital hemorrhage (arrow) grossly visible in a 5-week-old homozygous mutant fish just before death. Similar bleeding was not observed by blinded observers in heterozygous or wild-type clutchmates. Whole mount in situ hybridization with an antisense probe demonstrates localization of f5 expression to the liver in f5+/+ zebrafish at 5 dpf (E), but not in f5−/− mutants (F). (G) Total mRNA was prepared from pooled larvae (4 dpf, n = 21 for each genotype) followed by quantitative real-time PCR. Error bars represent standard deviation and statistical significance was determined by a Student t test.
Phenotypes of f5 homozygous mutants reveal adult lethality and spontaneous hemorrhage in homozygous mutant adult fish. (A) Survival curves of zebrafish offspring from f5+/− incrosses, starting at 2 months of age, demonstrate progressive loss of 90% of homozygotes by 7 mpf. Heterozygotes also demonstrate statistically significant decreased survival compared with wild-type fish, with 50% mortality by 1.5 years of age. (B) Genotype distributions of offspring from f5+/− incrosses evaluated at various stages demonstrate loss of homozygotes beginning at 28 dpf. (C) Spontaneous hemorrhage distal to the dorsal fin (arrow) in a 4-week-old homozygous mutant fish. (D) Severe spontaneous periorbital hemorrhage (arrow) grossly visible in a 5-week-old homozygous mutant fish just before death. Similar bleeding was not observed by blinded observers in heterozygous or wild-type clutchmates. Whole mount in situ hybridization with an antisense probe demonstrates localization of f5 expression to the liver in f5+/+ zebrafish at 5 dpf (E), but not in f5−/− mutants (F). (G) Total mRNA was prepared from pooled larvae (4 dpf, n = 21 for each genotype) followed by quantitative real-time PCR. Error bars represent standard deviation and statistical significance was determined by a Student t test.
Zebrafish embryos and larvae show no signs of overt hemorrhage
Loss of F5 in mice results in bimodal mortality with one-half of homozygous mutant embryos dying at embryonic days 9 and 10, and the remaining from massive hemorrhage within 2 hours of birth.10 Surprisingly, no appreciable loss of homozygous mutant fish embryos was observed until 28 dpf (Figure 2A). Given embryonic hemorrhage observed in the murine model, we carefully examined zebrafish at early stages of development. Larvae from heterozygous incrosses were stained with o-dianisidine at 7 dpf and examined for any evidence of hemorrhage. No differences in o-dianisidine staining was seen between homozygous mutants and their wild-type and heterozygous clutchmates (data not shown). Given the extended survival and lack of hemorrhage in early embryos and larvae, we hypothesized that there might be residual expression or altered localization of f5 mRNA in mutants. In zebrafish, the liver bud is first distinguishable at approximately 2 dpf, overlying the yolk sac to the left of midline.46 The liver grows to its final dimensions between 2.5 and 3 dpf with outgrowth complete by 5 dpf. Expression of f5 was examined in the developing zebrafish at 5 dpf, and we confirmed localization to the liver, but it was absent in f5−/− mutants (Figure 2E-F). These data are consistent with nonsense mediated decay, which has been demonstrated in other mutants.24,35 Reverse transcription PCR indicated reduction of f5 in heterozygotes and significantly decreased expression in homozygotes (Figure 2G). Given the early frameshift mutation, as well as lack of alternative products seen with cDNA amplification, any translated protein is predicted to be nonfunctional.
f5 homozygous mutants are unable to form thrombi in an induced model of venous and arterial endothelial injury
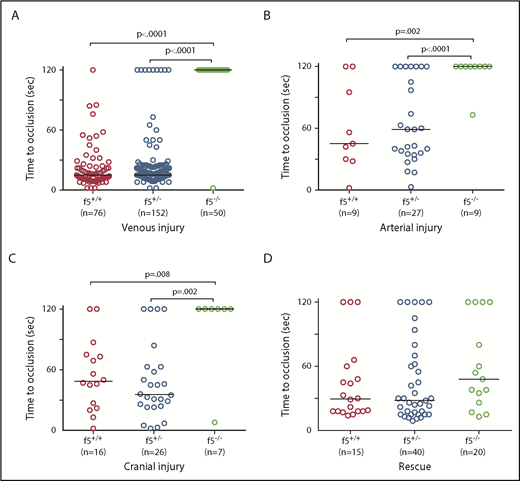
Because there was no spontaneous bleeding visible during the embryonic/larval period, we evaluated whether f5 mutants respond to induced trauma. We performed laser-mediated endothelial injury on larvae generated from f5+/− incrosses, targeting the PCV (orthologous to the mammalian inferior vena cava) at 3 dpf, the dorsal aorta (orthologous to the mammalian aorta) at 9 dpf, and the PMBC at 6 to 7 dpf. As expected, wild-type and f5+/− larvae exhibited complete vessel occlusion within 2 minutes, whereas f5−/− mutants failed to occlude (venous P ≤ .0001, arterial P = .002, PMBC P = .008 by the Mann-Whitney U test; Figure 3A-C). Interestingly, the f5+/− larvae exhibited a trend toward longer time to occlusion and increased numbers of larvae, which did not occlude compared with wild-type, but this was not statistically significant. To confirm the role of FV deficiency in this phenotype, as well as to rule out off target mutations generated through genome editing, we injected f5+/− incrosses with a zebrafish f5 cDNA under control of the zebrafish ubiquitin (ubi) promoter (pDestTol2pA2_ubi:f5-p2A-EGFP).47 Subsequent injury of the PCV at 3 dpf demonstrated significant occlusion, with rescue of the f5−/− phenotype in 73% of transgene-injected embryos (Figure 3D, P < .0001 by the Mann-Whitney U test).
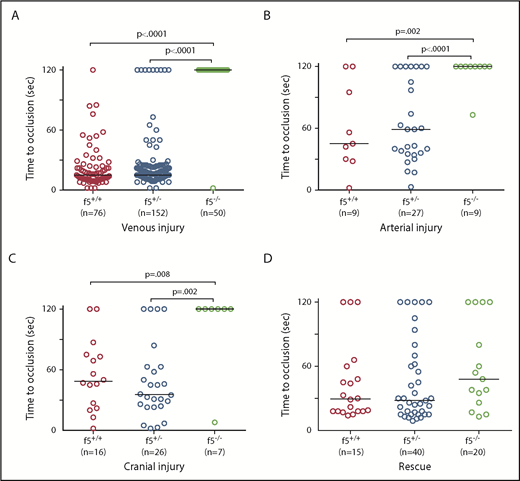
Effects of F5 deficiency on hemostasis in the venous and arterial circulation. (A) Laser-mediated endothelial injury of the PCV was performed on larvae at 3 dpf. The time to occlusion was significantly prolonged in f5−/− larvae in comparison with f5+/− and f5+/+ siblings (P < .0001, Mann-Whitney U test). (B-C) Endothelial injury was also performed on the dorsal aorta at 9 dpf, and PMBC at 6 to 7 dpf with similar results. (D) Injection of wild-type zebrafish f5 cDNA under control of the zebrafish ubiquitin promoter into 1-cell stage embryos resulted in significant rescue of the PCV hemostatic defect at 3 dpf in 73% of f5−/− larvae when compared with uninjected mutants (P < .001). Note that variability in the time to occlusion is a result of technical factors and heterozygosity in the genetic background. This has been seen with laser injury across other zebrafish mutants.
Effects of F5 deficiency on hemostasis in the venous and arterial circulation. (A) Laser-mediated endothelial injury of the PCV was performed on larvae at 3 dpf. The time to occlusion was significantly prolonged in f5−/− larvae in comparison with f5+/− and f5+/+ siblings (P < .0001, Mann-Whitney U test). (B-C) Endothelial injury was also performed on the dorsal aorta at 9 dpf, and PMBC at 6 to 7 dpf with similar results. (D) Injection of wild-type zebrafish f5 cDNA under control of the zebrafish ubiquitin promoter into 1-cell stage embryos resulted in significant rescue of the PCV hemostatic defect at 3 dpf in 73% of f5−/− larvae when compared with uninjected mutants (P < .001). Note that variability in the time to occlusion is a result of technical factors and heterozygosity in the genetic background. This has been seen with laser injury across other zebrafish mutants.
Inhibition of fibrinolysis has no significant effect on the f5−/− larval hemostatic defect
Given the delayed lethality of homozygous mutants as compared with the murine model, we suspected the possibility of alternate pathways for fibrin production. To investigate this, we evaluated whether treatment with an antifibrinolytic, ε-aminocaproic acid, could restore venous occlusion in homozygous mutants. Previously, we demonstrated ε-aminocaproic acid mediated reversal of the hemostasis defect in fibrinogen-deficient at3 mutants,24 but not in f10−/− larvae.35 In f5 homozygous mutants, laser-mediated endothelial injury revealed that treatment with ε-aminocaproic acid resulted in no statistically significant rescue (Figure 4).
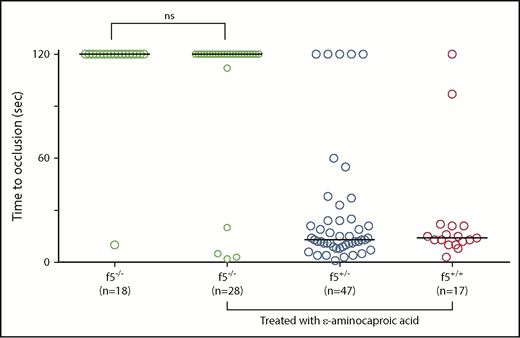
Antifibrinolytic treatment with ε-aminocaproic acid does not restore hemostasis in f5 mutants. Offspring from f5+/− incrosses were treated with ε-aminocaproic acid at 1 dpf and tested for venous occlusion in response to laser-mediated endothelial injury at 3 dpf, followed by genotyping. There was a slight increase in the percentage of f5−/− embryos that formed an occlusive thrombus (14.3% vs 5.5%) compared with untreated clutchmates, but this was not statistically significant. Horizontal bars represent the median time to occlusion. ns, not significant.
Antifibrinolytic treatment with ε-aminocaproic acid does not restore hemostasis in f5 mutants. Offspring from f5+/− incrosses were treated with ε-aminocaproic acid at 1 dpf and tested for venous occlusion in response to laser-mediated endothelial injury at 3 dpf, followed by genotyping. There was a slight increase in the percentage of f5−/− embryos that formed an occlusive thrombus (14.3% vs 5.5%) compared with untreated clutchmates, but this was not statistically significant. Horizontal bars represent the median time to occlusion. ns, not significant.
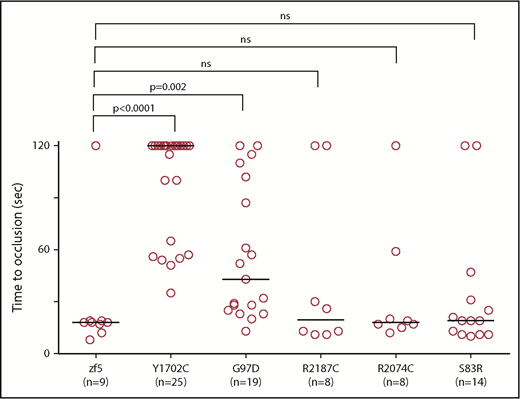
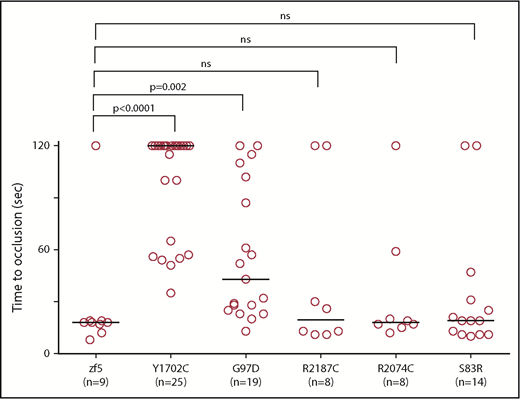
In vivo functional evaluation demonstrates variable severity of defects in VUS. Human F5 variants were engineered into the orthologous positions in the zebrafish f5 cDNA under the control the of the ubiquitin promoter. Expression vectors were injected into 1-cell stage embryos from f5 heterozygous incrosses. Laser-mediated endothelial injury was performed at 3 dpf and time to occlusion recorded, followed by genotyping. Although Y1702C and G97D demonstrated residual occlusion, they were unable to effectively rescue (P < .0001, P = .002, respectively, by the Mann-Whitney U test). Conversely, S83R, R2074C, and R2187C did not show statistically significant differences from wild-type rescue (not significant by the Mann-Whitney U test). Horizontal bars represent the median time to occlusion. Numbering reflects the human amino acid positions.
In vivo functional evaluation demonstrates variable severity of defects in VUS. Human F5 variants were engineered into the orthologous positions in the zebrafish f5 cDNA under the control the of the ubiquitin promoter. Expression vectors were injected into 1-cell stage embryos from f5 heterozygous incrosses. Laser-mediated endothelial injury was performed at 3 dpf and time to occlusion recorded, followed by genotyping. Although Y1702C and G97D demonstrated residual occlusion, they were unable to effectively rescue (P < .0001, P = .002, respectively, by the Mann-Whitney U test). Conversely, S83R, R2074C, and R2187C did not show statistically significant differences from wild-type rescue (not significant by the Mann-Whitney U test). Horizontal bars represent the median time to occlusion. Numbering reflects the human amino acid positions.
Evaluation of VUS in humans with F5 deficiency through in vivo evaluation in zebrafish f5−/− mutants
With widespread availability and decreasing costs of sequencing, increasing numbers of variants of unknow significance (VUS) are being identified. We previously demonstrated proof of concept for the use of zebrafish for this purpose and identified the causative mutations in patients with FX deficiency.35 We evaluated 5 F5 mutations that had been identified in patients with clinical diagnoses of FV deficiency (Table 2). All 5 of the affected amino acids are conserved across species; thus, we engineered the variants into the zebrafish f5 cDNA expression vector. These mutant plasmids were injected into zebrafish embryos from f5 heterozygous incrosses at the 1-cell stage, followed by laser-mediated endothelial injury at 3 dpf. Wild-type f5 expression plasmid, as well as the S83D, R2187C, and R2074C substitutions, were able to reverse the hemostatic defect. G97D demonstrated some activity, but was still significantly reduced from wild-type, whereas Y1702C showed almost no activity, correlating with the phenotypes observed in patients.
Discussion
We report targeted mutagenesis of zebrafish f5 using CRISPR/Cas9-mediated genome editing with development of a model of type I FV deficiency. Given early lethality resulting from hemorrhage in mice, we hypothesized that there would be significant bleeding during embryonic/larval stages; however, a large percentage of our homozygous mutants survived into adulthood. Embryos have a brief period of maternal RNA and protein expression, which could be hypothesized to allow longer term survival, but transcriptomic and proteomic data demonstrate that there is no FV expression during this period.48,49 In contrast to f10 mutants, which demonstrated a median survival of 1 to 2 mpf,35 f5 homozygotes demonstrated progressive loss with a median survival of 4 mpf. This might be expected given the role of FV as a cofactor in FX-mediated conversion of prothrombin to thrombin. Absence of a cofactor might be better tolerated than absence of the protease because the former condition could retain prothrombinase activity, albeit at significantly lower efficiency. In contrast to f10+/− mutants, fish heterozygous for the f5 mutation demonstrated decreased survival compared with their wild-type counterparts, which we hypothesize is due to a mild bleeding tendency that we found is present as early as 3 dpf.
This is the third example of long-term survival of a coagulation factor knockout in fish when compared with its murine counterpart. Previously, we demonstrated that genome editing of zebrafish at324 and f1035 both result in survival into adulthood, despite mid-gestation embryonic and/or neonatal lethality in mice. Examination of the f5−/− mutant larval vasculature, including arterial and venous vessels, demonstrated the inability to form thrombi in response to endothelial injury. This suggests a nearly complete hemostatic defect, so we speculate that the prolonged survival could be due to 1 or more of several mechanisms. Lethality in mice often occurs in the neonatal period, with bleeding occurring secondary to birth trauma. Yolk-based external, nonplacental development in the laboratory aquatic environment likely poses fewer hemostatic challenges than those encountered through the birthing process and terrestrial environs. Systemic blood pressures in mice are much higher than in zebrafish,50,51 and this may also play a role in differential risk of bleeding. Although the coagulation cascade is highly conserved between fish and mammals,30,52 it is possible that fish have evolved unique factors or pathways that enable greater tolerance of severe bleeding defects.
We have previously observed that the hemostatic defect in At3 coagulation factor–deficient larvae could be partially reversed by treatment with the fibrinolysis inhibitor ε-aminocaproic acid, but not in f10 mutants.24,35 These data suggest that in the former case there is enough residual fibrinogen and fibrin formation for there to be an effect, and the latter is consistent with what appears to be a complete block in fibrin production. We hypothesized that because FX is a protease and FV is a cofactor, we might see evidence for a low level of fibrin formation in the f5 mutants given that the protease is still present. This was supported by the longer term survival observed for the f5 homozygous mutants as compared with the f10 knockout. However, the results of fibrinolysis inhibition on induced thrombus formation were the same for both of these common pathway mutations. These data suggest that, at least in the short term of the endothelial injury assay, there is not enough fibrin generated to form an occlusive thrombus.
The ability to evaluate specific human mutations for in vivo phenotypic rescue is an invaluable tool for determining whether novel mutations are deleterious. Because the promise of whole genome and whole exome sequencing has become reality, increasing numbers of VUS have been discovered, leaving clinicians with difficulties when interpreting significance. Various databases and common resources are available to aid in this endeavor but do not provide a complete picture. In vivo evaluation of such mutations provides an opportunity for further characterization of the expected phenotype. Our ability to evaluate specific, clinically identified mutations is dependent on conservation across species, but this does not pose a significant hindrance because most deleterious mutations occur within well conserved regions.53 This evaluation is particularly useful within the field of coagulation where preanalytic variables and coincident coagulation perturbations can influence testing. Evaluation of VUS within the F5 locus can help to identify causative mutations and provide a more nuanced estimation of an individual’s patient’s bleeding phenotype. This is exceptionally advantageous in FV deficiency, in which factor levels do not correlate well with bleeding phenotype. Further evaluation of mutations in this way may help to provide explanations for the differing phenotypes seen between patients with similar factor levels.
We evaluated 5 mutations, 2 of which had previously been described and investigated, and 3 VUS that were predicted to be deleterious, all of which affect conserved residues from humans to zebrafish. Y1702C is a well-described missense mutation within the A3 domain that causes protein instability and may interfere with formation of disulfide bridges.54-56 Homozygosity results in severe FV deficiency (FV antigen of 1% and activity of <1%54 ) and no significant rescue was observed in our evaluation. G97D (A1 domain) and R2187C (C2 domain) have not been previously described but were found to be compound heterozygous in a patient with an FV antigen level of 10%. G97D was not able to restore venous occlusion in our analysis, although R2187C was able to do so, implying that the latter is a less deleterious mutation that may compensate clinically. S83R (A1 domain) has not previously been described and was found to be homozygous in a patient with FV antigen and activity levels of 23% and 4%, respectively. Despite its low activity, it was able to effectively restore venous occlusion. R2074C (also in the C2 domain) results in decreased secretion of FV and enhanced intracellular degradation, and homozygosity results in low but detectable FV activity (4%).57 Venous occlusion was restored despite this defect, demonstrating that this molecule does retain in vivo coagulant activity and strategies that mitigate the secretion defect could be exploited as a clinical strategy to restore hemostasis in affected patients. Because our assay is qualitative, rather than quantitative, lack of rescue could be due to either type I or II mutations (ie, reduced levels of FV antigen and/or activity).
All mutations demonstrated some ability to enhance occlusion, even when statistically different from wild-type FV, demonstrating that as in mammals, low levels of FV activity may be sufficient for augmentation of thrombin generation in zebrafish as assessed by our laser-induced endothelial injury model. This is consistent with the relatively mild clinical bleeding phenotypes observed, even in patients with unmeasurable FV activity. Half normal endogenous thrombin potential has been shown with as little as 1% FV, with absent or mild bleeding found in patients with endogenous thrombin potentials of 30% of higher.14 Although minimal levels are sufficient for thrombus formation in response to laser-induced endothelial injury, there is a nonsignificant trend of prolonged time to occlusion and/or total lack of occlusion in f5+/− zebrafish. We hypothesize that this mild bleeding tendency over time is what ultimately leads to the shortened survival seen in heterozygous fish compared with wild-type siblings.
Interestingly, our rescue results somewhat correspond with both bleeding symptomatology and reported factor activity levels. Although the model was able to accurately predict bleeding symptoms with the Y1702C mutation, it is likely that it would miss subtle phenotypes such as the bruising tendency described in the patient with the R2074C mutation. Historically, activity level has had limited correlation with severity of bleeding.58 Our in vivo evaluation could be beneficial in patients for whom factor activity levels do not correlate with bleeding phenotype, performed early on, and provide treatment and prognostic guidance. Ideally, we would have liked to evaluate mutations from patients with identical laboratory testing and divergent symptomatology. Unfortunately, given the limited number of patient mutations available, none met these conditions; this is a limitation to our assay. Because patients with identical activity levels may have quite different bleeding phenotypes, this tool may prove advantageous in providing a more nuanced view of individual hemostasis. The fecundity of zebrafish and ease of working with large numbers allow for easily repeated experiments, which may ultimately unveil hemostatic differences which would otherwise remain undiscovered.
There are several limitations to this approach. Primarily, despite the conservation of the overall protein and affected amino acids, there are still species-specific differences in other areas of the molecule. We are also unable to directly measure levels of FV activity in individual injected larvae and thus cannot quantitate the threshold level required for rescue. Another limitation is that the model does not currently take into account the FV platelet (thrombocytes in zebrafish) pool, or tissue factor pathway inhibitor levels. Although zebrafish thrombocytes do express FV,59 the ubi promoter drives ubiquitous expression, so we are unable to differentiate between effects within the plasma and platelet pools. However, for these studies, thrombocytes are not relevant as they do not appear until 5 to 6 dpf of life, whereas our studies of hemostasis were largely performed at 3 dpf. Future studies aim to address the role of these other facets of hemostasis.
In summary, our model indicates strong conservation between zebrafish and mammalian FV, including the site of its synthesis and necessity for hemostasis. Surprisingly, embryos and larvae tolerate what is a severe, lethal defect in mammals, allowing further studies not available in the mouse. This viability, now across 3 coagulation factor mutants with important roles in the common pathway, suggests the possibility of species-specific factors enabling survival. Further studies to identify such factors or other genetic modifiers could lead to novel diagnostic and therapeutic modalities for patients with hemostatic disorders.
Acknowledgments
This study was supported (in part) by research funding from a National Hemophilia Foundation/Shire Clinical Fellowship award; a Hemostasis and Thrombosis Research Society/Novo Nordisk Mentored Research Award in Hemophilia and Rare Bleeding Disorders from the Hemostasis and Thrombosis Research Society, which was supported by an educational grant from Novo Nordisk Inc.; grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (T32 HL007622) (A.C.W.), Eunice Kennedy Shriver National Institute of Child Health and Human Development (K12 HD028820) (A.C.W.), and National Heart, Lung, and Blood Institute (T32 HL125242) (S.J.G.); National Hemophilia Foundation Judith Graham Pool Award (M.S.R.); Ricerca Corrente of the Italian Ministry of Health (F.P.); National Institutes of Health, National Heart, Lung, and Blood Institute (grants R01 HL124232 and HL125774); and the Bayer Hemophilia Awards Program (J.A.S.). J.A.S. is a recipient of the American Society of Hematology Scholar Award and is the Diane and Larry Johnson Family Scholar of Pediatrics.
Authorship
Contribution: A.C.W. designed and performed research, analyzed data, and wrote the manuscript; S.J.G., K.I.L., and M.S.R. designed and performed research and analyzed data; A.C.F. and C.E.R. performed research and analyzed data; R.A., S.D., M.M., and F.P. designed research; R.A. and S.D. genetically characterized F5-deficient patients; and J.A.S. designed and supervised research, analyzed data, and edited the manuscript.
Conflict-of-interest disclosure: A.C.W. has received consulting fees from Shire and Kedrion. F.P. has received honoraria for participating as a speaker at satellite symposia and educational meetings organized by Ablynx, Grifols, Novo Nordisk, Roche, Shire, and Sobi; has received consulting fees from Kedrion; and is member of the scientific advisory board of Ablynx. J.A.S. has received consulting fees from Shire, Bayer, CSL Behring, and Octapharma. The remaining authors declare no competing financial interests.
Correspondence: Jordan A. Shavit, Department of Pediatrics, University of Michigan, Medical Science Research Building III, Room 8301, 1150 West Medical Center Dr, Ann Arbor, MI 48109; e-mail: jshavit@umich.edu.