

Abstract
Immune thrombotic thrombocytopenic purpura (iTTP) caused by an autoantibody-mediated deficiency of ADAMTS13 and atypical hemolytic syndrome (aHUS) caused by alternative complement dysregulation are the most common primary thrombotic microangiopathies (TMAs). The evaluation of a patient with TMA is a medical emergency since it is critical to quickly distinguish iTTP and aHUS from other causes of TMA. Untreated iTTP is rapidly fatal, and delays in initiating complement inhibition in aHUS increase the risk of irreversible renal failure. An ADAMTS13 activity level of less than 10% is diagnostic of iTTP in the appropriate clinical setting. In settings where rapid-turnaround ADAMTS13 testing is not available, clinical features and clinical prediction tools are useful to identify patients who should receive emergent plasma exchange. We present an evidence-based approach to the initial (first 24 hours) diagnosis and management of iTTP and review the clinical and laboratory features that can be used to identify patients with aHUS who will benefit from early C5 blockade. We also discuss the potential use of complement blockade to improve outcomes in selected patients with secondary TMA.
Learning Objectives
Identify patterns of clinical features and use clinical decision tools to distinguish iTTP from aHUS
Understand the interpretation and application of ADAMTS13 assays for iTTP diagnosis
Introduction
Thrombotic microangiopathies (TMAs) are a group of disorders characterized by thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and ischemic organ injury. The differential diagnosis includes primary TMA syndromes such as immune thrombotic thrombocytopenic purpura (iTTP) and atypical hemolytic uremic syndrome (aHUS, or primary complement-mediated TMA) and multiple causes of “secondary” TMA (Figure 1). While different TMAs have distinct, and sometimes overlapping, pathogenic mechanisms, their clinical presentations are similar and thus present a diagnostic challenge. Rapid and accurate initial diagnosis is critical because untreated iTTP is immediately life-threatening. Delays in initiating complement inhibition for aHUS also increase the risk of permanent renal failure. This review focuses on the critical first 24 hours in managing TMAs and centers on distinguishing iTTP or aHUS from other causes of TMA.
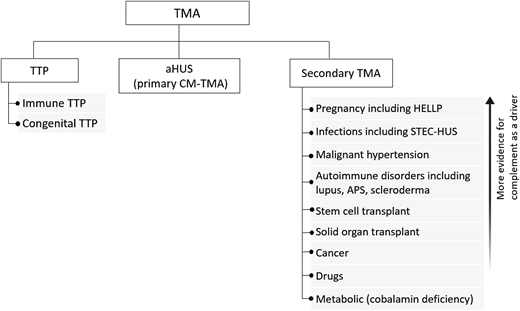
Practical approach to the differential diagnosis of TMA. The initial differential diagnosis of a TMA focuses on differentiating TTP from aHUS, also termed primary complement-mediated TMA, from the multiple causes of secondary TMA since this will impact initial treatment. There is evidence the complement acts as a driver for some secondary TMAs (with and without complement mutations), and complement inhibition may be a useful therapeutic strategy if control of the underlying disorder does not lead to improvement in the TMA and renal function. CM-TMA, complement-mediated TMA.
Practical approach to the differential diagnosis of TMA. The initial differential diagnosis of a TMA focuses on differentiating TTP from aHUS, also termed primary complement-mediated TMA, from the multiple causes of secondary TMA since this will impact initial treatment. There is evidence the complement acts as a driver for some secondary TMAs (with and without complement mutations), and complement inhibition may be a useful therapeutic strategy if control of the underlying disorder does not lead to improvement in the TMA and renal function. CM-TMA, complement-mediated TMA.
CLINICAL CASE
A 45-year-old woman presented to the emergency department after experiencing headache, malaise, and dark urine for 3 days. Physical examination showed an elevated blood pressure of 174/98 mmHg and mild abdominal tenderness. An initial laboratory evaluation showed a hemoglobin level of 8.5 g/dL, a platelet count of 32 × 109/L, a mean corpuscular volume of 89 fL, a creatinine level of 1.8 mg/dL, and a lactate dehydrogenase (LDH) level of 998 U/L. The peripheral smear showed thrombocytopenia and numerous schistocytes (Figure 2).
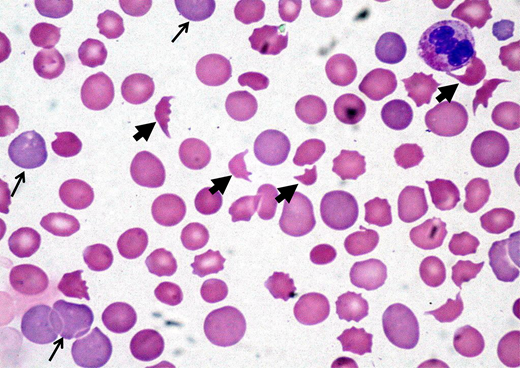
Peripheral blood film showing classic findings of TMA. The image shows features of thrombocytopenia and microangiopathic hemolysis with an increase in schistocytes (bold arrows). Reticulocytes (narrow arrows) are also increased and indicate increased red cell turnover.
Peripheral blood film showing classic findings of TMA. The image shows features of thrombocytopenia and microangiopathic hemolysis with an increase in schistocytes (bold arrows). Reticulocytes (narrow arrows) are also increased and indicate increased red cell turnover.
Initial evaluation of a patient with suspected TMA
The initial step in the evaluation of a patient with possible TMA is to confirm thrombocytopenia and MAHA (schistocytes). Notably, more than 1% schistocytes (correlating to >2 per high power field) is considered a robust indicator of TMA. Rarely, schistocytes may not be appreciably increased in patients with early iTTP, and we recommend examining another blood smear 12 to 24 hours later when the clinical picture is highly suspicious for TMA.1 The next step is to evaluate for causes of secondary TMA or conditions such as disseminated intravascular coagulation or severe cobalamin deficiency that can mimic TMA. Patients with recent diarrheal illness should be evaluated for Shiga toxin–producing Escherichia coli (STEC)-HUS.2 Table 1 summarizes the laboratory workup of a suspected TMA. The most time-sensitive decision is whether therapeutic plasma exchange (TPE) should be initiated emergently for possible iTTP. A secondary goal is to avoid unnecessary risks from central venous catheter insertion and plasma exchange and minimize delays in administering effective therapy with eculizumab or ravulizumab for aHUS.
Comparison of presenting clinical and laboratory findings in iTTP and aHUS
| Immune TTP6,7 | aHUS (adult)8,9,44 | |
|---|---|---|
| Adult or childhood onset | More common in adults (Congenital TTP more common in children) | Similar prevalence of adults and children |
| MAHA with thrombocytopenia | 100% | ~85% |
| Neurologic findings | Overall, 67%-78% Seizure, 7%-8% Coma, 3%-10% Stroke, 5%-10% | Overall, 8%-43% |
| Thrombocytopenia | More severe Median: 10-15 × 109/L | Less severe Median: 57-118 × 109/L |
| Renal dysfunction | Less severe Median serum creatinine: 1.09-1.3 mg/dL | More severe Median serum creatinine: 2.9-4.6 mg/dL |
| Severe (or malignant) hypertension | N/A, uncommon | Common, 8%-54% |
| Cardiac involvement | 42% | 19% |
| Fever | 10%-24% | Not reported |
| Abdominal symptoms | 35% | 15% |
| ADAMTS13 activity (%), median (range)16 | 5 (0-11) | 66.5 (12-119) |
| Immune TTP6,7 | aHUS (adult)8,9,44 | |
|---|---|---|
| Adult or childhood onset | More common in adults (Congenital TTP more common in children) | Similar prevalence of adults and children |
| MAHA with thrombocytopenia | 100% | ~85% |
| Neurologic findings | Overall, 67%-78% Seizure, 7%-8% Coma, 3%-10% Stroke, 5%-10% | Overall, 8%-43% |
| Thrombocytopenia | More severe Median: 10-15 × 109/L | Less severe Median: 57-118 × 109/L |
| Renal dysfunction | Less severe Median serum creatinine: 1.09-1.3 mg/dL | More severe Median serum creatinine: 2.9-4.6 mg/dL |
| Severe (or malignant) hypertension | N/A, uncommon | Common, 8%-54% |
| Cardiac involvement | 42% | 19% |
| Fever | 10%-24% | Not reported |
| Abdominal symptoms | 35% | 15% |
| ADAMTS13 activity (%), median (range)16 | 5 (0-11) | 66.5 (12-119) |
Pathophysiology of iTTP and aHUS
In iTTP, an autoantibody-mediated deficiency of ADAMTS13 causes the accumulation of ultralarge von Willebrand factor (VWF) multimers, leading to platelet aggregation and microvascular thrombi.3 In contrast, aHUS is caused by dysregulated (uncontrolled) alternative complement pathway activation that triggers inflammation, platelet activation, and direct endothelial injury. Over 50% of patients with primary aHUS have a genetic or acquired defect in complement regulation.4 aHUS is frequently triggered by a complement-amplifying condition such as infection, pregnancy, or surgery. TMA in the setting of a coexisting condition such as infection, autoimmune disease, cancer, pregnancy, stem cell or solid-organ transplant, or certain medications (tacrolimus, cytotoxic drugs) is often categorized as secondary TMA or secondary aHUS, in which the underlying condition is considered the cause rather than a “trigger.” Complement activation is implicated as a driver of some secondary TMAs associated with pregnancy (hemolysis, elevated liver enzymes, and low platelets, or HELLP syndrome), malignant hypertension, autoimmune disorders, and infections.5 It may be difficult to distinguish whether these conditions are a cause of secondary TMA or a trigger for aHUS.
iTTP vs aHUS: clinical features and clinical prediction tools
Though their clinical presentations are similar, certain features provide clues to the likelihood of iTTP vs aHUS. iTTP more often manifests neurologic symptoms and severe thrombocytopenia, but renal impairment is less common.6,7 In contrast, renal impairment is nearly universal in aHUS, and more than 20% exhibit extrarenal manifestations, including diffuse or focal neurologic events, cardiac injury, and gastrointestinal symptoms including diarrhea, vomiting, and pancreatitis (Table 2).8,9 New or worsening severe hypertension is a “red flag” for aHUS.8,9 A recent study reported pathogenic complement variants in over half of patients with malignant hypertension-associated TMA who had poor renal outcomes and did not respond to antihypertensives alone, suggesting that some malignant hypertension may actually be aHUS. While its distinction from malignant hypertension due to other causes is challenging, consider aHUS in young patients without known hypertension or TMA and renal failure that worsens despite blood pressure control.10
Laboratory evaluation of patients presenting with an acute TMA
| Tests | Indication | Utility |
|---|---|---|
| CBC, reticulocyte count; LDH; bilirubin (total, indirect), haptoglobin; blood smear | All patients | Establish diagnosis of TMA with thrombocytopenia, hemolytic anemia, and schistocytes. |
| PT, aPTT, D-dimer, fibrinogen | All patients | Rule out disseminated intravascular coagulation. |
| ALT, AST; creatinine, urinalysis, cardiac troponin, ECG | All patients | Assess organ damage. |
| ADAMTS13 activity | All patients | CONFIRM or rule out TTP. |
| ADAMTS13 inhibitor and antibody | If ADAMTS13 < 20% | Confirm immune vs congenital TTP. Consider ADAMTS13 sequencing if ADAMTS13 inhibitor and antibody tests are persistently negative and ADAMTS13 activity remains less than 20% in remission. |
| Stool culture/Shiga toxin | Recent diarrheal illness | Confirm STEC-HUS |
| Vitamin B12, MMA, homocysteine | Pancytopenia, suggestive blood smear (extreme pleomorphism, macro-ovalocytes, hypersegmented neutrophils | Assess for severe vitamin B12 deficiency that can present as “pseudo-TTP” |
| Autoimmune screening (eg, antiphospholipid antibodies, lupus anticoagulant, ANA) | aHUS with autoimmune history | Identify secondary TMA vs aHUS trigger |
| Infectious workup (eg, cultures, HIV) | Based on symptoms and history | Identify secondary TMA vs aHUS trigger |
| Renal biopsy | If cause of renal injury is unclear | Can distinguish TMA from other cases of renal injury and thrombocytopenia (eg, tubular necrosis from sepsis or medications, posttransplant rejection) but cannot distinguish specific etiology of renal TMA. |
| Tests | Indication | Utility |
|---|---|---|
| CBC, reticulocyte count; LDH; bilirubin (total, indirect), haptoglobin; blood smear | All patients | Establish diagnosis of TMA with thrombocytopenia, hemolytic anemia, and schistocytes. |
| PT, aPTT, D-dimer, fibrinogen | All patients | Rule out disseminated intravascular coagulation. |
| ALT, AST; creatinine, urinalysis, cardiac troponin, ECG | All patients | Assess organ damage. |
| ADAMTS13 activity | All patients | CONFIRM or rule out TTP. |
| ADAMTS13 inhibitor and antibody | If ADAMTS13 < 20% | Confirm immune vs congenital TTP. Consider ADAMTS13 sequencing if ADAMTS13 inhibitor and antibody tests are persistently negative and ADAMTS13 activity remains less than 20% in remission. |
| Stool culture/Shiga toxin | Recent diarrheal illness | Confirm STEC-HUS |
| Vitamin B12, MMA, homocysteine | Pancytopenia, suggestive blood smear (extreme pleomorphism, macro-ovalocytes, hypersegmented neutrophils | Assess for severe vitamin B12 deficiency that can present as “pseudo-TTP” |
| Autoimmune screening (eg, antiphospholipid antibodies, lupus anticoagulant, ANA) | aHUS with autoimmune history | Identify secondary TMA vs aHUS trigger |
| Infectious workup (eg, cultures, HIV) | Based on symptoms and history | Identify secondary TMA vs aHUS trigger |
| Renal biopsy | If cause of renal injury is unclear | Can distinguish TMA from other cases of renal injury and thrombocytopenia (eg, tubular necrosis from sepsis or medications, posttransplant rejection) but cannot distinguish specific etiology of renal TMA. |
ALT, alanine aminotransferase; ANA, antinuclear antibody; aPTT, activated partial thromboplastin time; AST, aspartate aminotransferase; CBC, complete blood count; ECG, electrocardiogram; MMA, methylmalonic acid; PT, prothrombin time.
Severely deficient ADAMTS13 activity is diagnostic of TTP. However, most centers do not perform ADAMTS13 testing in-house, and results of send-out tests can take up to 3 to 7 days. Clinical prediction models, such as the French TTP score and PLASMIC score, use readily available clinical features and laboratory testing to identify patients who should receive emergent TPE (Table 3).11,12 Importantly, these scores should be applied only to patients with TMA and cannot replace clinical judgment. The French score includes serum creatinine, platelet count, and antinuclear antibody,11 but the 2-component score with only the platelet count and serum creatinine is most commonly used. A score equal to or greater than 1 has a sensitivity of 98.8% for iTTP and a specificity of 48.1%.11 Thus, a French score of 0 is useful to rule out iTTP, but a score equal to or greater than 1 does not always rule in iTTP. The PLASMIC score has 7 components and classifies patients as low risk (score <5), intermediate risk (5), and high risk (6-7) for iTTP, and these categories correspond to a 0% to 4%, 5% to 25%, or 62% to 82% probability of iTTP, respectively. An intermediate or higher PLASMIC score (≥5) has 99% sensitivity and 57% specificity for iTTP,12,13 and this threshold is considered sufficient to start plasma exchange for presumed iTTP. An advantage of the PLASMIC score is that it has been validated in multiple other cohorts and as a meta-analysis of 13 studies.13 Compared to the French score, it has a higher C statistic (0.93 vs 0.88; P = .0032) and classifies fewer patients into the intermediate-risk group compared to the French score.12 Notably, the sensitivity of both the French and PLASMIC scores decreases to approximately 75% in older adults (>60 years) who are at increased risk of iTTP-related mortality but often present with atypical features such as worse renal function and less profound thrombocytopenia.14 These tools may also be less reliable in relapsed iTTP, which is often diagnosed early through patients' familiarity with symptoms or laboratory surveillance. Pregnancy-associated TMA represents a particularly high-risk situation, and these scores have not been specifically validated for pregnancy-associated and peripartum TTP. Neither score has been validated in pediatric populations and may require adjustment to incorporate representative measures of renal dysfunction across children of different age groups.
PLASMIC and French scores for iTTP
| Clinical prediction tool | Score components (1 point for each) | Probability of severe ADAMTS13 deficiency | ||
|---|---|---|---|---|
| Low | Intermediate | High | ||
| PLASMIC score | Platelet count <30 × 109/L Hemolysis: reticulocytes >2.5%, undetectable haptoglobin, or indirect bilirubin >2 mg/dL No active cancer No history of solid-organ or stem cell transplant MCV <90 fL INR <1.5 Creatinine <2 mg/dL | 0-4 | 5 PLASMIC score ≥5: sensitivity, 99%; specificity; 57%; NPV, 99%; PPV, 56%13 | 6-7 PLASMIC score ≥6: sensitivity, 85%; specificity, 89%; NPV, 92%; PPV, 81%)13 |
| French score | Platelet count <30 × 109/L Serum creatinine <2.26 mg/dL Positive ANAa | 0 | 1 French score ≥1: sensitivity 98.8%; specificity 48.1%; NPV, 93.3%; PPV, 85%11 | ≥2 French score 3; sensitivity 96.9%; specificity 98.1%; NPV, 38.6%; PPV, 98.7% |
| Clinical prediction tool | Score components (1 point for each) | Probability of severe ADAMTS13 deficiency | ||
|---|---|---|---|---|
| Low | Intermediate | High | ||
| PLASMIC score | Platelet count <30 × 109/L Hemolysis: reticulocytes >2.5%, undetectable haptoglobin, or indirect bilirubin >2 mg/dL No active cancer No history of solid-organ or stem cell transplant MCV <90 fL INR <1.5 Creatinine <2 mg/dL | 0-4 | 5 PLASMIC score ≥5: sensitivity, 99%; specificity; 57%; NPV, 99%; PPV, 56%13 | 6-7 PLASMIC score ≥6: sensitivity, 85%; specificity, 89%; NPV, 92%; PPV, 81%)13 |
| French score | Platelet count <30 × 109/L Serum creatinine <2.26 mg/dL Positive ANAa | 0 | 1 French score ≥1: sensitivity 98.8%; specificity 48.1%; NPV, 93.3%; PPV, 85%11 | ≥2 French score 3; sensitivity 96.9%; specificity 98.1%; NPV, 38.6%; PPV, 98.7% |
The modified French score that includes only platelet count and serum creatinine is more commonly used (since ANA is rarely available at presentation) and has similar performance metrics.
ANA, antinuclear antibody; INR, international normalized ratio; MCV, mean corpuscular volume.
Laboratory diagnosis of TTP
ADAMTS13 activity assays
Most ADAMTS13 activity assays are performed by fluorescence resonance energy transfer (FRET) or chromogenic enzyme-linked immunosorbent assay methods detecting cleavage of a VWF peptide (containing the ADAMTS13 cleavage site) by patient plasma.15 Similar to functional assays for coagulation factors, results are expressed as a percent of normal (pooled plasma) activity. ADAMTS13 activity greater than 10% is diagnostic of TTP, and levels lower than 20% are inconsistent with TTP.16 Borderline ADAMTS13 activity between 10% and 20% poses a diagnostic dilemma.17 In a report from the Oklahoma TTP registry, 3 of 78 patients with confirmed iTTP had ADAMTS13 activity of more than 10%, attributed to differences between ADAMTS13 assays or inhibitory anti-ADAMTS13 antibodies that dissociate in vitro, falsely elevating ADAMTS13 activity.6 ADAMTS13 activity of 10% to 20% may also be seen in iTTP samples collected after plasma transfusion. ADAMTS13 activity between 20% and 60% is common in patients with inflammatory disorders, sepsis, liver disease, and aHUS.16,18 Some patients with aHUS have ADAMTS13 in the 10% to 20% range.16,18 Thus, clinical judgment is critical when interpreting borderline ADAMTS13 activity. Patients with ADAMTS13 activity of 10% to 20% and a high probability of iTTP (based on clinical presentation and PLASMIC score) should be treated as having iTTP, especially if they are responding to TPE. Confirming an ADAMTS13 inhibitor or autoantibody supports the diagnosis of iTTP. However, if the clinical picture is atypical for iTTP (renal failure) and there is no improvement after 3 to 5 sessions of TPE, another diagnosis should be considered.
ADAMTS13 inhibitor and autoantibody assays
Most pathogenic anti-ADAMTS13 antibodies are immunoglobulin G, targeting the spacer domain of ADAMTS13, and have an inhibitory or neutralizing function that can be detected in a Bethesda-like assay.19,20 The ADAMTS13 inhibitor assay may be negative in patients with nonneutralizing antibodies that accelerate clearance.21 ADAMTS13 antibody testing detects both neutralizing and nonneutralizing antibodies and is more sensitive for iTTP. However, specificity is lower, as nonneutralizing ADAMTS13 antibodies are present in 4% of healthy individuals and 5% to 13% of those with autoimmune disorders.22
Rapid ADAMTS13 testing
Assay time, technical expertise, and the low cost-efficiency of running individual samples compromise the ability of most centers to obtain ADAMTS13 results in the time frame required to make clinical decisions (4-8 hours). Several “rapid” ADAMTS13 assays have been developed, including a semiquantitative screening assay (Technoclone) based on flow-through technology and an automated quantitative chemiluminescence-based immunoassay (HemosIL AcuStar).15 The semiquantitative Technoclone assay has a sensitivity of approximately 90%.23 While this assay may help with rapid screening, the sensitivity of 90% suggests that rigid adherence to the prescribed cutoff is not advised and that immediate confirmatory testing must be done in all cases, limiting its utility.23 In comparison, the HemosIL AcuStar assay provides results in approximately 30 minutes, correlates well with the standard FRET assay with a high level of agreement (kappa = 0.97) in identifying samples with severe ADAMTS13 deficiency (<10%), and appears to have discriminatory ability even below the limit of detection of the FRET assay.24,25 Wider adoption of rapid in-house ADAMTS13 testing is potentially the most impactful intervention to reduce health care costs while improving quality of care by rapidly identifying TTP and avoiding potentially harmful interventions.
Congenital TTP
It is usually impossible to distinguish between cTTP and iTTP at initial presentation, and all patients are treated with TPE and immunosuppression. Presentation in early childhood or a family history of iTTP may be clinical clues to cTTP, and the detection of ADAMTS13 inhibitor or autoantibody confirms iTTP. Patients without anti-ADAMTS13 inhibitor/antibody who do not recover ADAMTS13 activity during clinical remission should undergo sequencing to establish the diagnosis of cTTP (Figure 3).
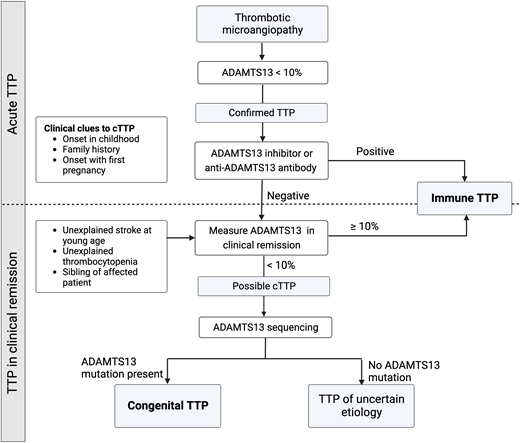
Diagnosing congenital TTP and distinguishing it from iTTP. While some clinical features, such as presentation early in life or a family history of TTP, may support a diagnosis of cTTP, it is virtually indistinguishable from iTTP during an acute episode. The presence of an inhibitor or antibody against ADAMTS13 confirms iTTP. Patients who do not recover ADAMTS13 activity levels over over 10% during clinical remission should undergo ADAMTS13 gene sequencing.
Diagnosing congenital TTP and distinguishing it from iTTP. While some clinical features, such as presentation early in life or a family history of TTP, may support a diagnosis of cTTP, it is virtually indistinguishable from iTTP during an acute episode. The presence of an inhibitor or antibody against ADAMTS13 confirms iTTP. Patients who do not recover ADAMTS13 activity levels over over 10% during clinical remission should undergo ADAMTS13 gene sequencing.
Laboratory diagnosis of aHUS
Complement-directed testing
Available complement testing cannot establish the diagnosis of aHUS, and treatment decisions must be based on clinical presentation and ruling out TTP, STEC-HUS, and secondary TMAs. We limit laboratory evaluation to tests that influence clinical management (Table 1). Serologic testing for complement proteins (eg, C3, C5, soluble C5b-9) or levels of factor H and I is available but has low sensitivity and specificity to identify aHUS.26 Additionally, variability across assays, effects of preanalytical handling, and significant overlap in levels between different TMAs and healthy individuals limit their use.27-29 Functional assays that rely on the in vitro deposition of C5b9 on endothelial cells or measure complement-dependent cell killing (modified Ham assay) appear to better discriminate between aHUS and non–complement- mediated TMA.30,31 However, these assays are not standardized and are currently available only in the research setting.
Sequencing to identify pathogenic variants in complement regulation genes and enzyme-linked immunosorbent assay for factor H autoantibody should be sent for patients with aHUS, but these results are usually not available for several weeks and do not affect the decision to start anticomplement therapy.
Genetic testing does impact long-term management by informing recurrence risk after renal transplant or discontinuing complement inhibition, and aids in counseling first-degree relatives.2 Complement mutations are found in 50% of aHUS as well as a significant proportion of patients with secondary TMAs, including malignant hypertension, HELLP syndrome, or catastrophic antiphospholipid syndrome.10 In the future, ultrafast genomic sequencing may aid the management of acute TMA, including the identification of rare, non–complement-mediated genetic disorders (eg, cobalamin C deficiency, DGKE mutations) and the confirmation of pathologic variants in complement genes.32 However, while genetic testing can help confirm a disgnosis of aHUS, it cannot be used to rule out a complement-mediated TMA.
Should I start plasma exchange or eculizumab urgently?
The decision to start TPE for likely iTTP vs up-front therapy with a complement inhibitor for suspected aHUS is influenced by the availability of rapid ADAMTS13 testing (Figure 4). When ADAMTS13 testing with a timely turnaround (4-8 hours) is not available, we recommend starting TPE and high-dose corticosteroids immediately in patients presenting with a TMA at intermediate or high risk for iTTP (PLASMIC score ≥5). If TPE is delayed due to a need for vascular catheter placement or transfer to a TPE-capable facility, we recommend transfusing plasma (10-15 mL/kg) and starting corticosteroids as a temporizing measure. For patients considered “low risk” for iTTP and worsening renal impairment, consider early initiation of anti-C5 therapy. The decision to continue TPE depends on ADAMTS13 results and clinical response. If ADAMTS13 activity is greater than 20%, TPE should be stopped since it does not improve outcomes in non-TTP TMAs, and we suggest starting a terminal complement inhibitor for likely aHUS. If renal function or clinical status worsens on TPE and ADAMTS13 results are not yet available, consider switching to anticomplement therapy for presumed aHUS.
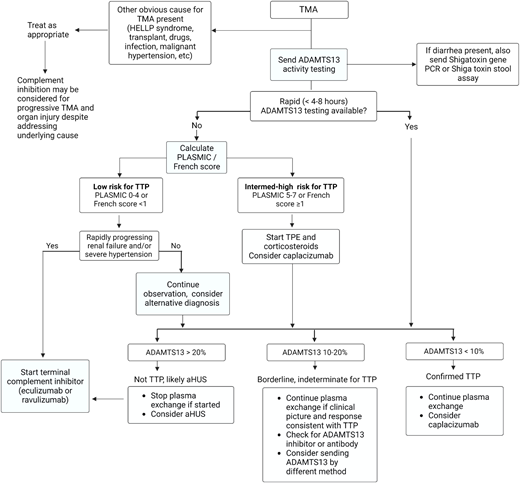
Approach to the initial evaluation and management of a patient with TMA. Early evaluation is focused on distinguishing iTTP or aHUS from secondary TMAs and minimizing delays in treating iTTP that is imminently life-threatening. The diagnostic pathway varied based on the availability of rapid-turnaround ADAMTS13 assays. PCR, polymerase chain reaction.
Approach to the initial evaluation and management of a patient with TMA. Early evaluation is focused on distinguishing iTTP or aHUS from secondary TMAs and minimizing delays in treating iTTP that is imminently life-threatening. The diagnostic pathway varied based on the availability of rapid-turnaround ADAMTS13 assays. PCR, polymerase chain reaction.
When available, rapid-turnaround ADAMTS13 testing can be used to distinguish between iTTP and aHUS. This reduces unnecessary TPE and delays in initiating effective therapy with eculizumab or ravulizumab in aHUS and facilitates the earlier use of targeted therapies (caplacizumab and rituximab) in iTTP.
Acute iTTP treatment—adjuncts to plasma exchange
In addition to plasma exchange, immunosuppression to suppress the anti-ADAMTS13 antibody is recommended. Most patients receive corticosteroids (usually prednisone at 1 mg/kg/d).17 Rituximab, an anti-CD20 monoclonal antibody that suppresses the production of anti-ADAMTS13 antibodies and reduces iTTP relapse, is also recommended for de novo and relapsed iTTP but should be started only after ADAMTS13 deficiency is confirmed. Caplacizumab is a nanobody directed against the A1 domain of VWF that inhibits VWF-dependent platelet aggregation and microthrombi formation.33 In the phase 3 HERCULES trial, caplacizumab along with TPE and immunosuppression led to faster resolution of thrombocytopenia and a 74% reduction in the composite end point of iTTP-related death, recurrence, and major thromboembolic events.33 Real-world experience also suggests that caplacizumab reduces refractoriness.34 The International Society on Thrombosis and Haemostasis guidelines on iTTP conditionally suggest using caplacizumab in acute iTTP (new diagnosis and relapsed disease) and starting caplacizumab even while waiting for ADAMTS13 testing since the benefit is likely maximized when started early.17 However, the rate of serious adverse events, particularly bleeding, is significantly increased with caplacizumab.17,33,35 Additionally, caplacizumab is not cost-effective based on acute iTTP outcomes at its current list price (approximately $270,000) in the United States.36 In contrast, a National Institute for Health and Care Excellence analysis from the United Kingdom accepted caplacizumab (with a different pricing agreement) as a cost-effective use of resources for a rare and highly morbid disease.37 None of these analyses were able to account for the potential impacts of caplacizumab on longer-term neurocognitive outcomes or of newer dosing strategies, which still need to be evaluated.38 Our practice is to start caplacizumab once ADAMTS13 at less than 10% is confirmed, outpatient availability of medication is assured, and bleeding risk is not increased (recent bleeding or antithrombotic therapy that cannot be stopped while on caplacizumab). For patients with severe neurologic symptoms, we consider starting caplacizumab before ADAMTS13 testing is completed. Platelet transfusions are associated with adverse outcomes, including mortality in iTTPn and should be used only for life-threatening hemorrhage.39
CLINICAL CASE (continued)
Our patient had an intermediate PLASMIC score of 5 and was started on TPE. Two days later, the platelet count was 38 × 109/L, but she developed anuric renal failure with a creatinine level of 3.8 mg/dL. TPE was stopped and eculizumab started, with improvement in her platelet count and hemolytic markers and, ultimately, the recovery of renal function. Five days after presentation, ADAMTS13 activity resulted as 56%.
Practical approach to the treatment of aHUS
Treatment with an anti-C5 monoclonal antibody is the standard of care for treating aHUS based on prospective nonrandomized trials that showed its clear benefit in improving hematologic TMA and renal function.2,40 The risk of end-stage renal disease and death has been reduced from 50% to 6% to 15% at 1 to 2 years.40 In the initial studies (trial 1, N = 17 and trial 2, N = 20) that led to the approval of eculizumab for aHUS, earlier treatment was associated with significantly greater improvement in glomerular filtration rate throughout the treatment period, though the median time to treatment initiation was several months (not days) in both trials.40 In extension studies, patients in trial 1 who were treated earlier showed ongoing improvement even after 1 year.41 In a recent pharmacoeconomic analysis, early (≤7 days) initiation of anti-C5 therapy is associated with 3.2 times lower odds of needing dialysis and lower health care utilization and cost.42 Both eculizumab and the longer-acting ravulizumab are approved for the treatment of aHUS. We prefer eculizumab for the initial treatment of aHUS because the cost of a therapeutic trial is lower in the setting of diagnostic uncertainty, where response to treatment often serves as the final diagnostic tool. Renal function recovery may be faster with eculizumab than ravulizumab, but this comparison is limited by differences in the cohorts in the studies evaluating eculizumab and ravulizumab. Patients in the ravulizumab cohort were older and had a lower rate of complement mutations.2
Distinguishing secondary aHUS from aHUS with a trigger is challenging, and the benefit of C5 blockade in most secondary aHUS is not unequivocally established. However, recent research has demonstrated a role of complement as a pathogenic mechanism or shown the efficacy of complement blockage in secondary TMAs due to autoimmune disorders (lupus and antiphospholipid syndrome), pregnancy, and some infections and drugs, including chemotherapeutics.5 For these secondary TMAs, we suggest eculizumab as a second-line therapy if addressing the underlying condition does not lead to improvement in the TMA and renal function.2,5 We suggest at least 2 weeks trial of therapy. Improvement in platelet count and LDH are expected within 10 to 14 days, although renal recovery is time dependent and may take months.40,43
Conclusion
Early and effective therapy for iTTP and aHUS drastically improves outcomes for these potentially devastating conditions. Currently, early treatment decisions must be made based on clinical features, tools such as the PLASMIC score, and ADAMTS13 testing, when available. Wider adoption of rapid ADAMTS13 testing will mitigate treatment delays, lower costs, and improve outcomes for both iTTP and aHUS. Functional complement assays and ultrarapid gene sequencing are being developed, which may help with diagnosing aHUS and predicting the response to complement inhibition in secondary TMAs. Improved diagnostic assays are critical to optimize the use of novel therapies in iTTP and aHUS.
Acknowledgments
This work was supported by the National Institutes of Health Heart, Lung and Blood Institute grant K99HL150594 (SC) and an American Society of Hematology Scholar Award (SC)
Conflict-of-interest disclosure
Charlotte M. Story: no competing financial interests to declare.
Gloria F. Gerber: advisory board: Apellis Pharmaceuticals, Alexion Pharmaceuticals; honorarium: Merck; stockholder (spouse): Pfizer.
Shruti Chaturvedi: advisory board: Alexion, Sanofi, UCB, Sobi, Takeda; consultancy: Alexion, Sanofi, UCB, Sobi, Takeda; honoraria/royalties: UpToDate.com, Dynamed.com.
Off-label drug use
Charlotte M. Story: All authors discuss the-off label use of rituximab for immune TTP, and eculizumab/ravulizumab for secondary TMA.
Gloria F. Gerber: All authors discuss the-off label use of rituximab for immune TTP, and eculizumab/ravulizumab for secondary TMA.
Shruti Chaturvedi: All authors discuss the-off label use of rituximab for immune TTP, and eculizumab/ravulizumab for secondary TMA.
