

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of excessive and maladaptive inflammation. Primary HLH is most frequently encountered in young children, and, without timely recognition and therapy, can lead to multiorgan failure and death. It is most often diagnosed using the HLH-2004 criteria and by identifying pathological mutations. However, the HLH-2004 criteria are not specific for HLH, and patients can easily fulfill these diagnostic criteria in other proinflammatory states in which HLH-therapy would not be indicated, including hematologic malignancies, infections, and rheumatologic disease. Therefore, great care must be taken to ensure that the specific disease associated with features of HLH is accurately recognized, as consequences of improper treatment can be catastrophic. We propose a diagnostic pathway for patients for whom HLH is on the differential (visual abstract). Importantly, in situations in which the initial diagnostic workup is equivocal or unrevealing, reevaluation for occult malignancy, infection, or rheumatologic disease would be prudent, as occult presentations may be missed on primary evaluation. Temporizing medications can be used in critically ill patients while awaiting secondary evaluation. By using this framework, clinicians will be able to more reliably discern primary HLH from other pro-inflammatory states and thus provide timely, appropriate disease-specific therapy.
Learning Objectives
Recognize the limitations of the current diagnostic paradigms for HLH, identifying the various pathologies that can manifest as HLH
Formulate diagnostic and therapeutic approaches for patients with suspected HLH
Hemophagocytic lymphohistiocytosis (HLH) is not a single disease; rather, the term describes a state of systemic inflammation that is disproportionate, maladaptive, or sometimes merely accompanying another disease.1,2 This state may arise from an inherited genetic lesion, from an acquired source of pathologic immune activation, or from a combination therein. Commonly used terminology has sought to distinguish between those cases attributed principally to genetic causes (which have been labeled as primary HLH) and those cases attributed largely to acquired causes of immune activation (which have been labeled as secondary HLH). This nomenclature may cause some confusion, as many cases likely arise from a combination of such inherited and acquired factors. Many of the genetic deficits which may precipitate or predispose a person to HLH affect perforin-mediated lymphocyte cytotoxicity, and this subset of primary HLH is often referred to as familial HLH (FHL).3-5 In such cases, the ensuing defective lymphocytes are vulnerable to uncontrolled activation, liberating cytokines such as interferon gamma, which, in turn, leads to macrophage activation. Additional genetic variants that activate lymphocytes and macrophages via different mechanisms (eg, XIAP deficiency), along with certain specific and increasingly recognized genetic mediators of granule-mediated cytotoxic dysfunction (eg, RAB27A, LYST), are grouped with FHL under primary HLH (Figure 1). Activated macrophages and cytokines can cause life-threatening organ dysfunction, making rapid recognition and treatment critical.6
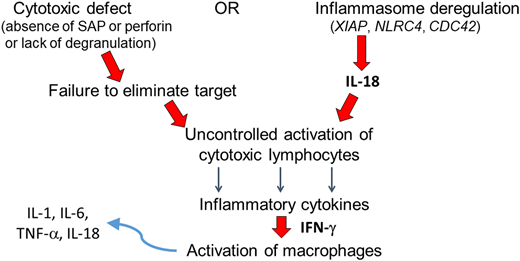
Pathogenesis of primary HLH and MAS. Genetic variants associated with primary HLH result in defective lymphocyte cytotoxicity (left pathway) or inflammasome degranulation, liberating IL-18 (right). Either mechanism culminates in uncontrolled activation of cytotoxic lymphocytes. The pathogenesis of MAS also involves IL-18-mediated activation of lymphocytes (all HLH patients may have elevated IL-18 levels; however, the increase may be particularly profound among those with MAS). Activated lymphocytes secrete inflammatory cytokines such as IFN-γ, activating macrophages. Activated macrophages in turn secrete additional inflammatory cytokines.
Pathogenesis of primary HLH and MAS. Genetic variants associated with primary HLH result in defective lymphocyte cytotoxicity (left pathway) or inflammasome degranulation, liberating IL-18 (right). Either mechanism culminates in uncontrolled activation of cytotoxic lymphocytes. The pathogenesis of MAS also involves IL-18-mediated activation of lymphocytes (all HLH patients may have elevated IL-18 levels; however, the increase may be particularly profound among those with MAS). Activated lymphocytes secrete inflammatory cytokines such as IFN-γ, activating macrophages. Activated macrophages in turn secrete additional inflammatory cytokines.
The Histiocyte Society led 2 clinical trials that improved the outcomes for children with primary HLH. The most recent trial, HLH-2004, enrolled patients aged 18 years and younger who either had genetically confirmed HLH or fulfilled 5 of 8 clinical criteria suggestive of HLH: fever, splenomegaly, cytopenia affecting at least 2 lineages, hyperferritinemia, hypofibrinogenemia or hypertriglyceridemia, hemophagocytosis in bone marrow or other tissue, reduced natural killer (NK)-cell cytotoxicity, or elevated sCD25 (sIL2Rα).7,8 These enrollment criteria were based on the experience and opinion of clinicians rather than on rigorous data. The sensitivity and specificity of these criteria for HLH remain unknown as there remains no gold standard for the diagnosis of HLH. However, the mere presence of features listed in these enrollment criteria has been adapted by much of the medical community as being diagnostic and pathognomonic of HLH.
When patients manifest these criteria in the absence of causal genetic defects, the condition is often called secondary HLH.1,9 Secondary HLH may be understood as an extraordinary inflammatory response to a proinflammatory trigger. Conditions which can lead to manifestations of secondary HLH are many and varied including infections, malignancies (most often lymphoma), and autoimmune disorders (including macrophage activation syndrome [MAS]). Although patients with all these conditions can also suffer life-threatening organ dysfunction, lumping these disparate pathologies under 1 umbrella term of secondary HLH is not scientifically justified.10 Treating patients with these varying diseases with therapies meant for primary HLH can lead to disastrous consequences.11 Making these distinctions at the bedside is a monumental challenge for clinicians. We present real-life cases to elucidate a practical approach to patients with suspected HLH.
CASE 1
A previously heathy 2-month-old female was brought to the emergency department with persistent fever and decreased oral intake. She was noted to be listless, febrile, hypoxic, tachycardic, and tachypneic and to have hepatosplenomegaly. Laboratory studies were notable for pancytopenia, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia (Table 1). She was resuscitated with intravenous fluids and treated with broad-spectrum antibiotics. Bone marrow biopsy showed normal hematopoiesis without increase in histiocytic or hemophagocytic forms.
Laboratory data
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Absolute neutrophil count ( × 103 per µL) | 0.14 | 0.89 | 1.9 | 0.08 |
| Hemoglobin (g/dL) | 7.4 | 8.7 | 7.8 | 8.1 |
| Platelet count ( × 103per µL) | 26 | 26 | 84 | 91 |
| Fibrinogen (mg/dL) | 50 | 163 → 75 | 759 | 167 |
| Ferritin (ng/mL) | 7000 | 18 000 | 4512 | >99 000 |
| Triglycerides (mg/dL) | NR | 153 | 192 | 178 |
| sCD25 (units/mL) | 88 228 | 71 439 | 34 750 | 1907 |
| Uric acid (mg/dL) | NR | 9.1 | 4.3 | NR |
| LDH (units/L) | NR | 1783 | 273 | 494 |
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Absolute neutrophil count ( × 103 per µL) | 0.14 | 0.89 | 1.9 | 0.08 |
| Hemoglobin (g/dL) | 7.4 | 8.7 | 7.8 | 8.1 |
| Platelet count ( × 103per µL) | 26 | 26 | 84 | 91 |
| Fibrinogen (mg/dL) | 50 | 163 → 75 | 759 | 167 |
| Ferritin (ng/mL) | 7000 | 18 000 | 4512 | >99 000 |
| Triglycerides (mg/dL) | NR | 153 | 192 | 178 |
| sCD25 (units/mL) | 88 228 | 71 439 | 34 750 | 1907 |
| Uric acid (mg/dL) | NR | 9.1 | 4.3 | NR |
| LDH (units/L) | NR | 1783 | 273 | 494 |
NR, not reported.
CASE 2
A previously healthy 2-year-old boy was brought to the emergency department with symptoms of fever, abdominal distension, and reduced oral intake. An evaluation revealed fever, tachycardia, pallor, hepatomegaly, and splenomegaly. Laboratory studies revealed pancytopenia, hyperuricemia, and hyperferritinemia (Table 1). He was treated with intravenous fluids and antibiotics. Over the next hours, fibrinogen decreased and urine output declined, requiring continuous renal replacement therapy. Bone marrow morphology showed prominent hemophagocytosis but otherwise normal hematopoiesis.
These 2 cases exemplify urgent life-threatening situations wherein simple tools such as the HLH-2004 criteria rapidly draw HLH into the differential diagnosis. Subsequent results showed elevated sCD25 levels in both children (Table 1), further raising the concern for HLH. Given the young age and absence of leukemia, primary HLH was considered most likely in both cases. The first child (2-month-old female) was treated with dexamethasone and etoposide and rapidly improved. Genetic studies revealed her to be homozygous for a variant of unknown significance in the PRF1 gene. Flow cytometry on peripheral blood lymphocytes showed absent perforin protein expression in NK and cytotoxic T lymphocytes (Figure 2), confirming the diagnosis of primary HLH caused by perforin deficiency. This illustrates the utility and power of flow cytometry assays (flow) in diagnosing primary HLH. In all cases of suspected HLH, we use flow for perforin and CD107a degranulation, the latter assessing the integrity of the lytic-granule secretory pathway.12 Normal degranulation depends on several proteins, many of which are encoded by genes implicated in primary HLH: UNC13D, STX11, STXBP2, LYST, RAB27A, and AP3B1.12 In males, we also add flow for SAP and XIAP. The biggest advantages of these assays are rapid turnaround time and precision. Moreover, CD107a degranulation is a functional assay, and the combination of perforin flow and CD107a has been shown to be more reliable than the NK-cell cytotoxicity assay to diagnose primary HLH.13 These assays are also helpful in situations wherein the results of genetic tests are equivocal, as above.
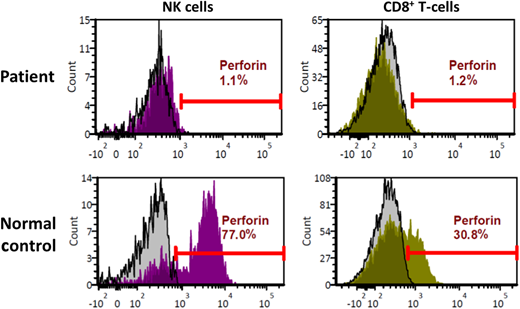
Perforin deficiency detected by flow cytometry. Histogram plots depicting flow cytometry analysis of peripheral blood lymphocytes from the patient described in case 1. Upper panels depict patient cells, while the lower panels depict normal controls. Results for natural killer (NK) cells are shown on the left, while those for CD8+ T lymphocytes are shown on the right. Isotype controls are represented by the gray histograms, and perforin antibody detection is represented by the histograms in magenta (NK) or green (CD8+).
Perforin deficiency detected by flow cytometry. Histogram plots depicting flow cytometry analysis of peripheral blood lymphocytes from the patient described in case 1. Upper panels depict patient cells, while the lower panels depict normal controls. Results for natural killer (NK) cells are shown on the left, while those for CD8+ T lymphocytes are shown on the right. Isotype controls are represented by the gray histograms, and perforin antibody detection is represented by the histograms in magenta (NK) or green (CD8+).
The second child (2-year-old), in addition to meeting the HLH-2004 criteria, also had hyperuricemia and rapidly developed renal failure, features not typical of primary HLH. His bone marrow aspirate and biopsy did not show leukemia, and full body CT scans only showed the known hepatomegaly and splenomegaly. Strikingly, quantitative polymerase chain reaction (PCR) for Epstein–Barr virus (EBV) on his peripheral blood demonstrated 25 × 106 copies/mL. His bone marrow biopsy showed an expanded population of CD8+ T lymphocytes, most of which were also positive for EBV-encoded-RNA, thus establishing the diagnosis of systemic EBV-positive T-cell lymphoma of childhood.14 Quantitative PCR on sorted lymphocytes showed the burden of T-lymphocyte EBV to be 700-fold and 23-fold higher than that associated with B lymphocytes or NK cells, respectively. His condition did not improve with dexamethasone treatment per the HLH-94 protocol. With the EBV results, treatment with multiagent chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) was initiated, and he rapidly recovered. All genetic and flow studies for primary HLH were normal. Following another cycle of chemotherapy with the same agents, he underwent allogeneic bone marrow transplant and is thriving. The association of EBV infection with HLH is well known.15 In primary HLH, EBV infection is recognized as a recurrent trigger of disease activation. There are other rare immunex deficiencies associated with severe EBV infection manifesting clinical features of HLH.16 In all these situations, EBV is associated with B lymphocytes. On the other hand, EBV infection of NK or T lymphocytes causes a spectrum of lymphoproliferative disorders (LPDs), from chronic smoldering mononucleosis-like illness (chronic active EBV) to fulminant lymphoma as in the second patient all without any identifiable genetic defects.14 Interestingly, EBV-driven NK- and T-cell LPDs are more common in people of East Asian and Indigenous American ethnicities. All these disorders can manifest clinical features fulfilling HLH-2004 and are frequently referred to as EBV-HLH or secondary HLH due to EBV.15,17 This broad term includes a disparate array of B- and NK/T-cell disorders, which are distinct from primary HLH, and require differing therapies. For example, treatment with the B-lymphocyte-directed anti-CD20 monoclonal antibody rituximab is beneficial in B-lymphocyte EBV disorders but not in T- or NK-cell LPDs.18 Rapid recognition and intervention with correct multiagent chemotherapy are crucial in fulminant NK/T-cell lymphomas. It is thus imperative to determine the specific condition in each patient.
CASE 3
A previously healthy 23-year-old developed persistent fever and fatigue. Investigations for infections were negative. Fevers persisted and laboratory studies revealed cytopenia and elevated liver enzymes. Bone marrow biopsy showed a nonspecific small population (5%) of atypical lymphocytes but was otherwise normal. Imaging studies demonstrated hepatosplenomegaly and lymphadenopathy. Lymph node needle-biopsy showed no malignancy or infection. With elevated sCD25 and ferritin (Table 1), he was diagnosed with HLH and treated with dexamethasone and intravenous immunoglobulin (IVIG). Symptoms promptly resolved but as dexamethasone was tapered, fever and fatigue recurred. He was then treated with dexamethasone and etoposide per HLH-94, with resolution of symptoms. Genetic studies for HLH revealed no variants. As treatment was weaned again, fever, fatigue, and cytopenia recurred, and he was referred for bone marrow transplant. His family requested a second opinion. In a search for illnesses that can mimic HLH, we performed a positron emission tomography scan showing innumerable fluorodeoxyglucose-avid osseous lesions and lymph nodes. An excisional lymph node biopsy revealed classic Hodgkin lymphoma. Chemotherapy for Hodgkin lymphoma was initiated with prompt resolution of all clinical signs of disease.
CASE 4
A 4-year-old boy was admitted to the hospital with fever, vomiting, bloody diarrhea, and dehydration. While hospitalized, he developed hepatosplenomegaly, pancytopenia, elevated ferritin, and triglycerides (Table 1). Bone marrow aspiration demonstrated hemophagocytosis with increased histiocytes. Having fulfilled criteria for HLH, therapy was initiated with dexamethasone and anakinra. The fever resolved, but signs of liver failure appeared with hyperbilirubinemia and coagulopathy. Additional therapy with etoposide was planned, but his family requested a second opinion. Given the bloody diarrhea and vomiting, we suspected an infection, and investigations revealed significant adenoviremia with a viral load of >200 × 106 copies/mL. Adenovirus-specific T-cell therapy was given, but his condition continued to worsen with rapid development of multiorgan failure; he died shortly thereafter. Postmortem examination determined the cause of death as disseminated adenovirus infection resulting in multiorgan failure. No genetic mutations were identified.
Cases 3 and 4 highlight the nonspecific nature of the HLH-2004 criteria, which resulted in unfortunate delays in correct diagnoses and potentially contributed to the death of the fourth patient. These occurrences are common and now account for the majority of referrals to specialized centers for patients with challenging or treatment-refractory HLH (Figure 3).11 The HLH-2004 criteria were created as the eligibility requirement for the clinical trial HLH-2004, and their specificity for primary HLH has never been shown. All the features listed in the criteria can be seen in many systemic inflammatory conditions, such as disseminated infections or malignancies like lymphoma. The specificity of these criteria for primary HLH is likely low.10 Contrary to popular belief and despite the eponym, hemophagocytosis is neither specific nor sensitive for primary HLH. Additionally, the NK-cytotoxicity assay also has poor specificity.13 One of the goals of the HLH-2004 guidelines was to utilize tests readily available at most centers.7 While these criteria and the subsequent HScore have certainly helped move HLH toward the forefront of clinicians' minds, our experience shows that in the diagnosis of primary HLH, these tools are strikingly nonspecific.10,11 The problem is not simply one of semantics or nomenclature. The prevailing dogma is that as soon as the diagnosis of HLH is reached, based on these nonspecific tools, it signals an urgent need for therapy with potent immunosuppresive agents such as high-dose corticosteroids, closely followed by etoposide. Such algorithmic decision-making often leads to inappropriate application of primary HLH–directed therapies in conditions in which such treatments may cause harm. Furthermore, the HLH-2004 trial was restricted to children up to the age of 18.8 There are no data on the outcome of applying such criteria to adults.19 The term secondary HLH is not a specific diagnosis but rather an amorphous, catchall description that can be applied to a variety of diseases. Since malignancies like lymphoma (as well as many of the other potential triggers of secondary HLH) are more common in adults compared to children, we suspect that majority of adults evaluated for primary HLH are likely manifesting other acute inflammatory diseases. Additional confusion arises when patients known to have an infection or lymphoma are also diagnosed with HLH on the basis of fulfillment of the HLH-2004 criteria or the HScore. Clinicians then face the conundrum of which condition to treat first—HLH or the trigger disease. In the vast majority of such situations, the label of HLH is attached simply because nonspecific criteria have been fulfilled, and HLH-directed therapy with steroids and etoposide is not needed. In the case of infections, such therapy is actually contraindicated. Rather, treatment should be directed at the primary pathology. In certain malignancies, liver dysfunction precludes chemotherapy. Many of these illnesses also fulfill the HLH criteria, and these patients are then often treated with steroids, sometimes with etoposide, resulting in improved liver function. While such a sequence could be interpreted as malignancy-associated HLH with response to HLH-therapy, steroids and etoposide are also efficacious in treating the vast majority of hematologic malignancies, and abatement of the cancer would then result in resolution of HLH.11 A recent study highlighted a subset of patients with hematologic malignancies associated with higher levels of ferritin and sCD25 and higher risk of mortality.20 It remains unclear if inflammation is directly implicated in the worse outcomes, or if these are biomarkers of treatment-refractory malignancies.
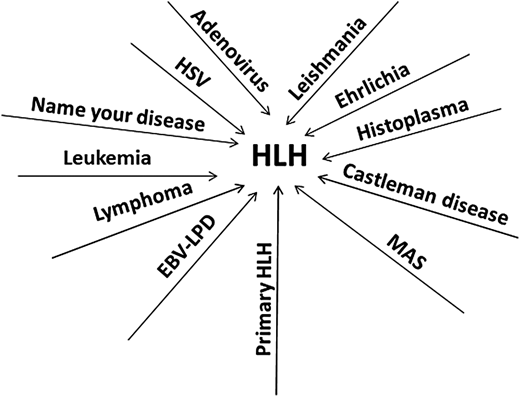
Many diseases can manifest clinical features of HLH. Depicted are the diseases we have seen as mistaken for and treated as primary HLH, akin to the many proverbial roads leading to Rome. HSV, herpes simplex virus.
Many diseases can manifest clinical features of HLH. Depicted are the diseases we have seen as mistaken for and treated as primary HLH, akin to the many proverbial roads leading to Rome. HSV, herpes simplex virus.
Discussion: our diagnostic approach
Many patients with a wide variety of proinflammatory illnesses may fulfill the HLH-2004 criteria or the HScore.10 The fulfillment of these criteria is not sufficient to diagnose HLH nor to initiate HLH-directed therapies.11 Awareness of the nonspecific nature of these tools and of the base rates of primary HLH (rare), infections, and lymphomas (common) are key components of accurate diagnosis. While testing for primary HLH should be pursued in the appropriate context, all cases of suspected HLH must also be examined for potential alternative diagnoses, including malignancies, infections, and rheumatologic disease. Broad HLH-directed testing includes assessment of ferritin, fibrinogen, sCD25, CXCL9, and IL-18, but clinicians must recognize that none of these tests is particularly specific for HLH.21 However, normal ferritin, normal sCD25, normal CXCL9, or elevated fibrinogen make HLH less likely. Evaluation for primary HLH includes flow for perforin, CD107a, SAP, and XIAP (the last two for males) and a comprehensive gene panel for IEIs or WES.12,22 While results are being awaited, ongoing assessment for other potential causes of hyperinflammation are prudent, such as bone marrow examination for leukemia positron emission tomography and computed tomography (and subsequent tissue biopsy if indicated) for lymphoma, and cultures and PCR/antigen assays for infections. Withholding steroids and immune suppression until biopsies have been obtained and infections ruled out should be strongly considered. Empiric treatment with antimicrobials should be pursued until infectious etiologies are sufficiently excluded. Certainly, if malignancy, infection, or other specific etiology is identified, the corresponding treatment should promptly be instituted.11 The finding of substantially high IL-18 may suggest MAS or an inflammasome disorder, and appropriate rheumatologic diagnostics may then be indicated.23 Abnormal CD107a, perforin, SAP, or XIAP expression makes familial HLH likely.12,22 These findings should be confirmed via genetic testing, and treatment should be considered with HLH-directed therapy (such as HLH-94 and anticytokine therapy) as warranted based on the clinical condition. If all these tests fail to yield a specific diagnosis, patients should be thoroughly reevaluated for occult malignancy, infection, or rheumatologic process. At the same time, whole exome sequencing may be considered. If a patient is clinically deteriorating while the workup is in progress and diagnosis remains uncertain, temporizing therapies including glucocorticoids, anticytokine agents, and/or IVIG may be considered.24-27 It is critical to recognize that most patients demonstrating HLH physiology will eventually be found to have an occult inciting condition (be it malignant, infectious, or rheumatologic) upon further evaluation. It should also be noted that many such cases may have multiple factors contributing to excessive inflammation and that even presentations of primary HLH may have an acquired trigger; therefore, even when primary HLH is identified, a search for triggering conditions should be undertaken. Although the past 2 decades have seen a tremendous increase in the awareness of HLH, they have also seen confusion regarding its accurate diagnosis and appropriate treatment. Although current screening tools, including the HLH-2004 criteria and H-Score, are diagnostically sensitive, they are also highly nonspecific, and these criteria may be fulfilled by many other severe illnesses. Evaluating suspected cases of HLH requires thoroughly investigating for other more common causes of excessive inflammation (including malignancy or infection) and using more specific testing for primary HLH (including flow cytometric and genetic testing). Such an approach may avoid missed diagnoses and inappropriate treatment of potential HLH mimics.
Disclosures
Ashish Kumar is a consultant for SOBI, Springworks therapeutics, and OPNA.
Eily Cournoyer: no competing financial interests to declare.
Leonard Naymagon: no competing financial interests to declare.
Off-label drug use
Ashish Kumar: There is nothing to disclose.
Eily Cournoyer: There is nothing to disclose.
Leonard Naymagon: There is nothing to disclose.
