

Abstract
Acute promyelocytic leukemia (APL), a phenotypically and genotypically unique subtype of acute myeloid leukemia, has seen unprecedented advances in its management since the introduction of all-trans retinoic acid (ATRA) and arsenic trioxide. However, the phenomenal pharmacologic conversion of this once highly fatal disease to one with a long-term survival exceeding 90% among patients who survive induction remains impaired by the significant incidence of early death (ED) reaching 30% in some real-world studies. The key driver for ED in APL is catastrophic hemorrhage with a proclivity for cranial sites. Most EDs in APL are currently considered preventable. Here, we discuss the concept of early death in APL and its characteristics. Importantly, we outline implementable strategies to reduce the incidence of ED. Early recognition of APL underpins these preventive measures as significant delays in the diagnosis increase the likelihood of ED. While early administration of ATRA is often taught to all hematology trainees, this lifesaving intervention is only possible if providers, including those in emergency departments and urgent/immediate care settings, are trained to have a high index of suspicion and competence to recognize the morphologic and clinical characteristics of the disease. Other proposed strategies tackle the complications that can be present at diagnosis or arise during induction therapy and address the issues of expert consultation and protocol adherence in the management of these patients. While some of these measures appear intuitive and others aspirational, widespread adoption could bring about an era of cure for almost every patient with APL.
Learning Objectives
Describe the concept of early death in acute promyelocytic leukemia (APL), its characteristics, and its key drivers
Propose strategies to prevent early death in APL
CLINICAL CASE
A 32-year-old man with a 2-week history of headaches and easy bruising sought medical attention in the emergency department. Complete blood count showed a white blood cell count of 60 000/µL, hemoglobin of 5.9 g/dL, and platelet count of 14 000/µL. The prothrombin was 22.8 seconds, activated partial prothrombin time 32.2 seconds, and fibrinogen 60 mg/dL. He received packed red blood cells, platelets, and cryoprecipitate units. The peripheral blood smear was suggestive of acute promyelocytic leukemia (APL). All-trans retinoic acid (ATRA) was started. A computed tomography scan of the head showed a small subarachnoid hemorrhage. A bone marrow aspiration/biopsy was consistent with the diagnosis of APL. Cytoreduction was initiated with hydroxyurea. Peripheral blood testing for PML::RARα returned positive, and arsenic trioxide and gemtuzumab ozogamicin were added to ATRA. The coagulopathy was managed with supportive transfusions. However, dyspnea and fever developed 7 days into his treatment. Examination showed edematous lower extremities and weight gain of 5 kg since admission. Oxygen saturation was 88% on ambient air, and he required 2 L of supplemental oxygen. Dexamethasone was administered at 10 mg twice daily for differentiation syndrome, and APL therapy was continued. There was improvement within 36 hours, and supplemental oxygen was weaned.
Introduction: Distinguishing features of APL
APL is the most curable subtype of acute myeloid leukemia (AML) in adults. The morphologic features of the leukemia cell are unique; the molecular pathogenesis has been deciphered; the clinical manifestations, primarily life-threatening bleeding, are characteristic; and the treatment is completely different from all other subtypes of AML.1 The disease is associated with a t(15;17) (q24;q21), resulting in the formation of the PML::RARα fusion transcript that blocks differentiation. Unlike other subtypes of AML in which leukemia cell genetics drive prognosis, the initial white blood cell (WBC) count at presentation is the most important prognostic factor in APL. The biologic basis for the relationship between a high WBC and prognosis is enigmatic. The WBC count is likely a surrogate for an as yet unidentified genetic marker. Almost miraculously, the vitamin A derivative ATRA and arsenic trioxide (ATO) induce differentiation and apoptosis of the promyelocytic leukemia cells. When administered together in low-risk patients without chemotherapy or in high-risk patients with either an anthracycline or the immunoconjugate gemtuzumab ozogamicin (GO), approximately 98% and 90%, respectively, are cured of their disease.2-6 This is provided that the patient survives induction since there is essentially no primary resistance in the ATRA-ATO era. Although cure in AML always depends on surviving induction, this is a particularly important concept in patients with APL since the major clinical manifestation, and one of the most distinguishing features, is a unique, complex, and potentially fatal bleeding diathesis. In fact, the major cause of treatment failure is not resistant disease, as is true of all other subtypes of AML, but rather early death (ED). ED in the context of APL is defined as death occurring within the first 30 days of diagnosis (although some studies define ED as within 30 days of admission or start of induction).7 Avoiding ED in patients with APL has emerged as the last frontier in the cure of all patients.
Early death in APL: Scope of the problem
Hemorrhage was recognized as a dominant feature of APL in the earliest report by Hillestad8 in 1957 that established APL as a distinct clinical entity. This description highlighted key characteristics: rapidly fatal course of a few weeks' duration, severe bleeding tendency due to fibrinolysis and thrombocytopenia, and normal erythrocyte sedimentation rate (ESR) probably due to reduced plasma fibrinogen. Now, more than 60 years after these observations, bleeding remains the major cause of ED and therefore the major cause of treatment failure.
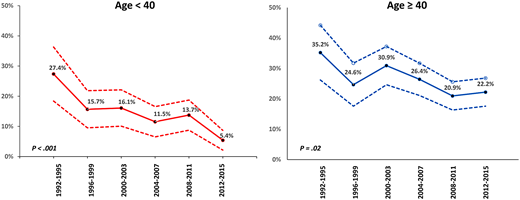
Historically, in large cooperative group trials of newly diagnosed patients treated with ATRA plus chemotherapy, the ED rate is between 5% and 10%, but the percentage of EDs attributable to bleeding is between 30% and 70%.9-13 However, these numbers reflect the outcome of younger patients generally without comorbidities who survive long enough to complete administrative requirements for enrollment on a clinical trial. In population-based studies, the ED rate is considerably higher and varies between 15% and 30%.7,14-17 The Swedish Acute Leukemia Registry reported an ED rate of 13% in younger patients (aged <35 years) but 25% overall with no change from 1997 to 2013.7 However, Jamy and colleagues16 reported significant progress, particularly in younger patients. Analysis of the Surveillance, Epidemiology, and End Results Registry revealed that ED rates have decreased significantly over time in younger adults, with a reduction in the ED rate from 27.4% between 1992 and 1995 to 5.4% between 2012 and 2015, but remained high in patients aged >40 years in whom the ED rate decreased only modestly from 35.2% between 1992 and 1995 to 22.2% between 2012 and 2015 (Figure 1). This is important since 30% of patients in the Swedish registry were older than 65 years.7 Bewersdorf and colleagues17 found that the inpatient mortality rate was 14% among 1464 admissions. Furthermore, 86% of low-risk patients were treated in accordance with National Comprehensive Cancer Network (NCCN) guidelines, whereas only 64.6% of high-risk patients were treated as recommended (Figure 2). This interesting observation may set the stage for a strategy to decrease the ED rate discussed below. The contribution of “delayed or missed diagnosis” to the ED rate is difficult to quantify as even population-based reports do not capture the patients who die early in emergency departments before a diagnosis is made. However, early identification of patients with APL must be a priority as it is key to the prevention of ED.
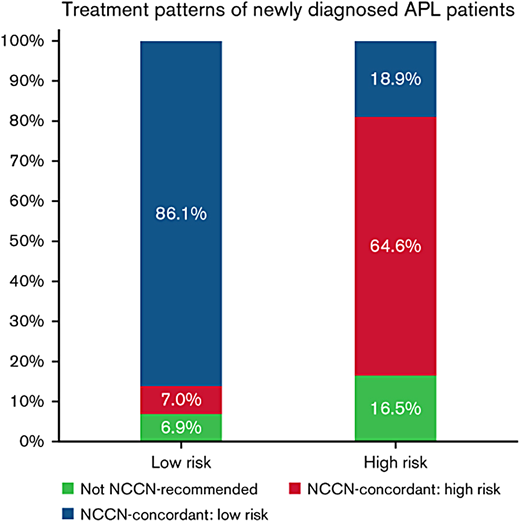
Treatment patterns of newly diagnosed patients with APL by risk group based on concordance with the NCCN guidelines.17
Treatment patterns of newly diagnosed patients with APL by risk group based on concordance with the NCCN guidelines.17
Characteristics of early death in APL
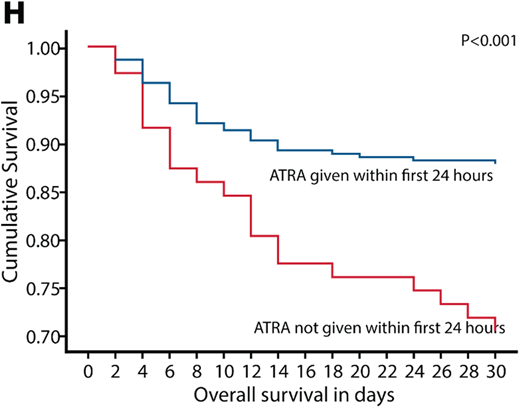
In a contemporary series, Gill et al18 reported an ED rate of 15.6% with ATRA administered ≥24 hours after presentation 19.8% of the time. Those patients for whom ATRA was administered within the first 24 hours had a better outcome than those receiving ATRA >24 hours from admission (Figure 3).18 This is an important observation since ED most commonly occurs within the first 4 days, particularly in the first 24 hours, and almost all EDs occur within 2 weeks.7,18 Bleeding is by far the most frequent cause of ED in APL. The Swedish registry reported 46% of EDs were attributable to bleeding.7 While intracranial hemorrhage is the most common presentation, life-threatening hemorrhage also can occur in the gastrointestinal tract and lungs.7 Other less common causes of ED include thrombosis, differentiation syndrome (DS), and infection.18 However, bleeding should be considered the most preventable cause of ED. Sepsis is the most commonly identified nonhemorrhagic cause of death in patients with APL, as demonstrated by the Swedish registry, which found it responsible for 15% of observed mortality.7 While it may seem counterintuitive, thrombosis is an often-unrecognized cause of morbidity and ED in APL.19,20 The pathogenesis of this apparent paradox is explained below. DS is a cardiorespiratory distress syndrome caused by ATRA, ATO, or both.21 The effect of differentiating agents such as ATRA and ATO on blast cells and promyelocytes results in increased production of proinflammatory cytokines (including tumor necrosis factor α and interleukins 1, 6, and 8), increased adhesivity of the blasts, and release of cathepsin G.22 These clinically manifest as systemic inflammation (fever, hypotension, tachycardia, and tachypnea) and increased vascular permeability with endothelial injury (pulmonary edema, peripheral edema, and weight gain).23 If unchecked, progression to multiorgan failure and death can result. However, with either prophylactic or early institution of corticosteroids, the syndrome may be less frequently problematic with very low mortality rates.
Factors predictive of ED have been reported. In addition to delays in ATRA administration, Gill et al18 reported that hypofibrinogenemia, WBC count ≥10 000/µL, and male sex predicted ED. The concept of high-risk disease (presenting WBC count ≥10 000/µL) should not be confused with high risk for ED as the former is designed to predict the risk for relapse following frontline therapy and is a marker for overall worse outcome. Bewersdorf and coworkers17 found that patient age, high-risk disease, and NCCN guideline nonconformant treatment were associated with higher odds of death or discharge to hospice. In a multivariate analysis of 5 clinical trials, WBC count >20 000/µL was the only independent predictor of hemorrhagic ED.24 The Swedish registry reported the highest rate of ED among patients with a WBC count in the 5000 to 10 000/µL range.7 Österroos and colleagues15 observed a linear increase in risk of ED with either age ≥50 years or WBC count ≥2200/µL. Importantly, there was an increased risk of ED even in patients with leukopenia. They divided age, WBC counts, and platelet counts into subgroups with assigned points to develop a risk score. Other groups have developed similar scoring systems.25 However, such scoring systems have limited practical value and have not influenced treatment strategies. Since ED may occur in patients with low-risk disease, the use of a different treatment approach in this group vs those with high-risk disease does not appear to be justified.
Pathogenesis of the coagulopathy: Bleeding and thrombosis
APL causes distinctive changes in the coagulation and anticoagulation systems, resulting in a coagulopathic state that can manifest as hemorrhage, thrombosis, or both (Figure 4).26 It has been proposed that APL causes bleeding by mechanisms that include primary and secondary fibrinolysis. Primary fibrinolysis is mediated by increased expression of annexin II and plasminogen activators (PAs) by malignant promyelocytes, resulting in increased levels of plasmin and consequently hypofibrinogenemia and elevated fibrin split products.27,28 Simultaneously, there is a decrease in the levels of circulating α2-antiplasmin, a potent antifibrinolytic protein, resulting in unchecked fibrinolysis. Secondary fibrinolysis is driven by an increased production of endothelial tissue PAs.29 Intracranial hemorrhage is also mediated by increased endothelial expression of annexin II and effects of increased levels of PAs in the brain.30 Podoplanin, an aberrantly expressed surface protein, contributes to thrombocytopenia and bleeding by promoting platelet aggregation and consumption; ATRA downregulates the expression of podoplanin and decreases risk of bleeding.31 Proteolysis of procoagulant proteins is another proposed mechanism.27 It was initially thought that the coagulopathy in APL is merely disseminated intravascular coagulation. However, fibrinolysis and proteolysis also play integral roles. APL appears less characterized by the formation of microvascular thrombi, and there are more marked derangements in coagulation parameters such as prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen, fibrin split products, and D-dimer while levels of anticoagulant proteins, such as proteins C and S, are preserved.32
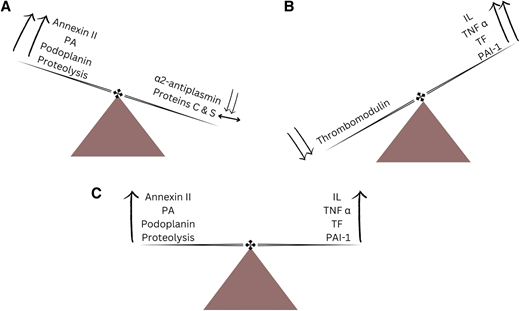
The dynamic interplay between the procoagulant and anticoagulant agents/mechanisms resulting in (A) hemorrhage, (B) thrombosis, and (C) concomitant hemorrhage and thrombosis. IL, interleukin; PAI-1, plasminogen activator inhibitor 1; TF, tissue factor; TNFα, tumor necrosis factor α.
The dynamic interplay between the procoagulant and anticoagulant agents/mechanisms resulting in (A) hemorrhage, (B) thrombosis, and (C) concomitant hemorrhage and thrombosis. IL, interleukin; PAI-1, plasminogen activator inhibitor 1; TF, tissue factor; TNFα, tumor necrosis factor α.
Thrombosis also can be a manifestation of disordered coagulation in APL, albeit far less commonly compared to hemorrhage.19 This may be attributable to hypercoagulability promoted by cytokines such as interleukins and tumor necrosis factor α, which enhance the activity of tissue factor and PA inhibitor 1 while decreasing the production of the endothelial anticoagulant thrombomodulin.26
Treatment of high-risk APL: Focus on prevention of early death
The modern treatment of APL is anchored on aggressive management of the coagulopathy and early initiation of ATRA. Differentiating agents, ATRA and ATO, have become the backbone of the treatment of APL.
Patients with high-risk APL should be treated according to one of several protocols for high-risk disease, initially aimed at controlling the WBC count and therefore perhaps decreasing the risk of ED. One approach is the addition of an anthracycline to ATRA and ATO. The APML4 study, which incorporates idarubicin in the induction phase of therapy on days 2, 4, 6, and 8 with ATRA and ATO, as well as ATRA and ATO in consolidation for 2 cycles and maintenance, results in 5-year disease-free survival and overall survival rates of 95% and 87%, respectively.4 An alternative strategy is the addition of GO, 9 mg/m2, given on day 1 of induction therapy, to ATRA and ATO in high-risk patients, which results in a 5-year disease-free survival and overall survival of 89% and 86%, respectively.2 Other clinical trials have also shown the benefit of adding GO in this patient population.11,33
Our approach to the management of patients with high-risk APL involves the combination of ATRA and ATO with GO. Consolidation therapy is given with 4 courses of ATRA and ATO without maintenance. The addition of an anthracycline, often idarubicin, is a very acceptable alternative if this is the clinician's preference or GO is unavailable. The use of GO may preserve cardiac function and decrease long-term cardiac toxicity, desired goals in both younger and older patients.34
In addition to early initiation of ATRA, proactive management of coagulopathy is vital and should be considered as important as ATRA. Due to the high risk of bleeding, there is essentially no role for prophylactic or therapeutic anticoagulation (including low-dose unfractionated heparin, which has historically been used in this patient population), and it should be avoided except possibly in the very rare setting of life-threatening thrombosis, an event the authors have never had to confront.
Although the benefit is not clearly established, we administer prophylactic steroids to all patients with APL given the minimal toxicities and would have done so in the clinical case presented. The choice of steroid and dose should be as administered in the regimen being followed. In patients managed without prophylactic steroids but who develop DS, the use of dexamethasone 10 mg every 12 hours until resolution has become the standard of care. Considering the risk of ED from infectious complications, especially in older adults treated with chemotherapy, the immunologic risks associated with use of prophylactic steroids should be carefully weighed against their anticipated benefit by the consultant hematologists and other experts involved in the care of these patients, particularly those with established infections.
The patient in the case described achieved complete remission with induction therapy and as anticipated had a favorable long-term outcome. The prompt initiation of ATRA and aggressive use of blood products to manage the coagulopathy likely prevented progression of intracranial hemorrhage. Prompt recognition of DS allowed early initiation of dexamethasone and allowed uninterrupted treatment with ATRA and ATO.
How to avoid early death in APL: A practical and aspirational guide
Given the natural history of the disease, there will always be some patients who come to medical attention with catastrophic bleeding whose fatal outcome cannot be prevented. However, for most patients whose outcome can be influenced, the following measures, some intuitive and others aspirational, are a guide to reduce the ED rate (visual abstract).
Practical measures
Promptly identify patients with APL as early recognition is key. Characteristic features include large ecchymoses, epistaxis, oral mucosal bleeding, and heavy menstrual bleeding. It is not usual for patients to report oral bleeding while brushing their teeth or flossing. The bleeding is often out of proportion to the degree of thrombocytopenia. It is important that the emergency department—often the first-contact health care professionals—reflexively obtain peripheral blood smear and coagulation profile (ie, PT, aPTT, D-dimer, and fibrinogen) for patients with an abnormal WBC count (typically neutropenia and/or blast cells) and/or thrombocytopenia. Every hematologist should be familiar with the unique morphologic features of APL on the peripheral blood smear (Figure 5).
Administer ATRA at the very earliest and slightest suspicion of APL in the emergency department with no delay for transfer to the inpatient floor or for pathology or genetic confirmation of the disease. Exposure to several days of ATRA is associated with very little or no toxicity if the diagnosis is not APL.
Be aggressive in blood product (cryoprecipitate and platelet units) support for the coagulopathy with cryoprecipitate and platelets with the goal to maintain the fibrinogen ≥150 mg/dL and platelet count ≥50 000/µL. Admittedly, both thresholds are arbitrary. Coagulation studies (ie, PT, aPTT, D-dimer, and fibrinogen) can be checked 2 to 3 times a day for the first 3 to 4 days.
Be vigilant for the detection of thrombosis. Clinicians should have a low threshold for imaging if there is a suspicion for thrombosis. Avoid the prophylactic use of anticoagulation.
Administer steroids in an effort to prevent or ameliorate DS. Use caution in patients with active infections.
Follow written guidelines (such as NCCN) for management in consultation with an expert in the field for day-to-day care, particularly early in the disease course. Choose 1 published regimen and follow it through without “mixing and matching” regimens to achieve optimal outcomes. In 2004, the American Society of Hematology established the International Consortium on Acute Promyelocytic Leukemia with its purpose to improve outcomes of APL in developing countries, including Brazil, through a uniform clinical protocol and establishment of a network for communication and collaboration.35 The success of this approach has been demonstrated. Historically in Brazil, the ED rate in APL was 32%.36 After the adoption of a uniform clinical protocol, the ED rate decreased to 15% in the participating countries.36
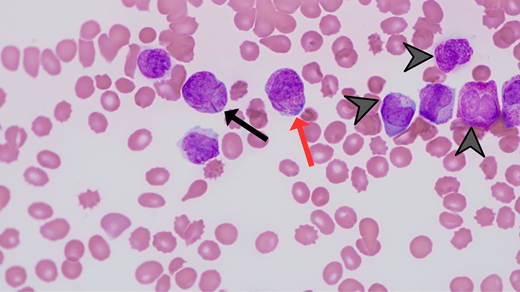
Photomicrograph (magnification 60 × ) depicting morphologic features of APL, including abundant primary azurophilic granules (red arrow), multiple Auer rods (black arrow), and reniform or bilobed nuclear contours (arrowheads).
Photomicrograph (magnification 60 × ) depicting morphologic features of APL, including abundant primary azurophilic granules (red arrow), multiple Auer rods (black arrow), and reniform or bilobed nuclear contours (arrowheads).
Aspirational measures
Emergency medicine training should include teaching about APL and its presentation clinical and laboratory characteristics. The curriculum should emphasize the key role of emergency medicine physicians in the prevention of ED, and trainees should achieve competency in early recognition of patients with the disease.
Emergency department management algorithms should be updated to include reflexive and STAT orders in patients for whom complete blood count reveals blast cells, abnormal neutrophils, and/or platelet counts.
Hospital standard operating procedures should ensure ATRA is immediately available. While many facilities see few patients with APL and therefore believe that maintaining stock is not cost-effective and a prohibitive administrative burden, they should establish a network with the nearest hospital that keeps ATRA on the shelf.
Although ED rates may improve over time without change in treatment approach, we recommend clinicians seek immediate consultation with a colleague with particular expertise in the disease. Jillella and colleagues37 have implemented a simple yet very effective strategy. A network of 7 experts across the United States available any time with brief, written, and practical management guidelines was established. Treatment was based on well-recognized standards of care with dose modifications for older adults and those with comorbidities. A decrease in the ED rate to 3.5% was observed, a level comparable to that observed in the recent Surveillance, Epidemiology, and End Results rates in younger patients, and this strategy serves as a paradigm that could be widely adopted as a way to provide free telemedicine physician-to-physician consultations regarding the care of patients with APL who are not already being treated by experts.
Acknowledgments
We thank Simbiat Odetola for assistance in the design of several figures and Dr Emily Alvey for the photomicrographs.
Conflict-of-interest disclosure
Oluwatobi Odetola: no competing financial interests to declare.
Martin S. Tallman: no competing financial interests to declare.
Off-label drug use
Oluwatobi Odetola: 6-mercaptopurine and methotrexate as maintenance in some protocols.
Martin S. Tallman: 6-mercaptopurine and methotrexate as maintenance in some protocols.
