

Abstract
Acquired hemophilia A (AHA) is an autoimmune disorder characterized by the formation of autoantibodies that neutralize the function of coagulation factor VIII. Immunosuppressive therapy (IST) with glucocorticoids, cyclophosphamide, rituximab, or combinations thereof is the standard of care to suppress autoantibody formation and induce remission of AHA. About 80% of patients achieve remission over the course of a few weeks to several months. However, patients with AHA are often elderly and frail and have adverse events from IST. Therefore, guidelines suggest an individualized approach using caution in elderly and frail patients. Prophylaxis with emicizumab may reduce the need for early and aggressive IST in the future.
Learning Objectives
Review guidelines on the use of immunosuppression in acquired hemophilia A
Discuss new concepts for reduced intensity or delayed immunosuppression
CLINICAL CASE
A 75-year-old woman is admitted for anemia and an extended forearm muscle bleed after venipuncture. During workup, her activated partial thromboplastin time is prolonged to 90 seconds. Factor VIII (FVIII) activity is reduced to <1% of normal, and acquired hemophilia A (AHA) is confirmed by the Nijmegen-modified Bethesda assay demonstrating an FVIII inhibitor of 28 BU/mL. Her previous medical history includes rheumatoid arthritis, arterial hypertension, and chronic obstructive pulmonary disorder; her concomitant medications include prednisolone 7.5 mg per day, methotrexate 10 mg per week, ramipril, and hydrochlorothiazide. Her general condition is compromised, with a World Health Organization (WHO) performance status of 3. She receives recombinant factor VIIa for 5 days to control the muscle bleed. A decision is made to postpone immunosuppressive therapy (IST) until her general condition has improved, and she receives prophylaxis with emicizumab. She receives outpatient care in the Hemophilia Care Center on a weekly basis and starts IST 4 weeks later, consisting of dexamethasone 40 mg per os (PO) (days 1-4 and 15-18) and rituximab 375 mg/m2 intravenous (IV) (days 1, 8, 15, and 22). FVIII activity slowly improves, and remission is achieved after 6 weeks.
Introduction
AHA is a severe bleeding disorder caused by autoantibodies that neutralize the activity of coagulation FVIII. Anti-FVIII autoantibodies, also known as “inhibitors,” can develop in previously healthy individuals, regardless of age or sex.1,2 Two peaks in AHA incidence are typically observed: one associated with pregnancy and another one with advanced age, typically >65 years. Of patients with AHA, 30% to 50% have underlying disorders, most commonly autoimmune disorders or malignancy.
Patients with AHA often have comorbidities and medications, such as antithrombotic agents or immunosuppressants, and require an individualized therapeutic approach. In contrast to congenital hemophilia, comparative clinical studies are not available in AHA, largely as a result of the rarity of the disorder. Care decisions are often based on the clinical experience of treating physicians, and expert center referral is recommended to provide the best possible care.
Traditionally, the therapeutic approach to AHA includes 2 distinct goals, one being the immediate control of acute bleeding through hemostatic medication, while the other is directed toward long-term eradication of autoantibody inhibitors through IST.3
Rationale for immunosuppressive therapy
The autoimmune disorder in AHA is polyclonal, as evidenced by the presence of anti-FVIII antibodies of different affinities, immunoglobulin classes and subtypes, and FVIII epitopes.4,5 Most patients also have autoantibodies against targets other than FVIII, suggesting a general disturbance of immune regulation.6 Monoclonal gammopathy, which is a typical condition associated with the acquired von Willebrand syndrome and is often unresponsive to standard immunosuppressive regimens, is rarely seen as an underlying disorder of AHA.
The indication for autoantibody eradication with IST is based on historic observational studies that have documented inhibitor- related mortality rates of 22% to 64%.7-9 More recent registry studies reported low mortality rates from bleeding (3%-9%) in patients receiving IST.10-14 However, the same studies reported that complications of IST, in particular infections, have become a leading cause of death among patients today. Therefore, recent guidelines suggested caution when using immunosuppressive therapy in elderly and frail patients.2
Goals of IST and definition of remission
The goal of IST is to reduce the risk of future bleeding by inducing remission of AHA. Spontaneous remission has been observed in patients not treated with IST, but this outcome is rare and unpredictable.15 Definitions of remission vary across studies and registries. The UK surveillance study defined complete remission (CR) as FVIII normal, inhibitor undetectable, and immunosuppression stopped or reduced to doses used before AHA developed without relapse.10 The GTH-AH 01/2010 study also included a definition for partial remission (PR): FVIII restored to >50% and no bleeding after stopping any hemostatic treatment for at least 24 hours.12
Guidelines on the use of IST in AHA
Two comprehensive guidelines have been published in the past 5 years.1,2 Both appeared in the preemicizumab era, when IST was the only way to protect patients from future bleeding episodes.
First-line options suggested were glucocorticoids alone or in combination with either cyclophosphamide or rituximab. Upfront combination therapy was suggested for patients with FVIII <1% or inhibitor titers >20 BU/mL, who were observed to need longer times to PR (median, 5-6 weeks) compared to patients with higher FVIII or lower inhibitor titers (3-4 weeks) in the observational GTH-AH 01/2010 study.12 The CREHA study compared glucocorticoids with cyclophosphamide against glucocorticoids with rituximab in first-line therapy of AHA, but results have not been published at the time of this review.16
Second-line IST is suggested if no decline in the inhibitor titer or rise in the baseline FVIII level is seen after 3 to 5 weeks of first-line therapy. Options include adding cyclophosphamide or rituximab, whichever was not used during first-line therapy, or alternative agents such as mycophenolate mofetil.17
Dosing recommendations for glucocorticoids, cyclophosphamide, mycophenolate mofetil, and rituximab are provided in Table 1.
Immunosuppressive drugs used to treat AHA
| Class or drug | 2019 guideline recommended dosing2 | Alternative dosing (CyDRi)20 |
|---|---|---|
| Glucocorticoids | Prednisolone or prednisone 1 mg/kg/d PO for a maximum of 4-6 weeks (followed by tapered withdrawal) | Dexamethasone 40 mg PO on days 1, 8, 15, and 22 |
| Cyclophosphamide | 1.5-2 mg/kg/d PO for a maximum of 6 weeks | 1000 mg IV on days 1 and 22 |
| Rituximab | 375 mg/m2 IV weekly for a maximum of 4 cycles | 100 mg IV on days 1, 8, 15, and 22 |
| Mycophenolate mofetil | 1 g/d for 1 week, followed by 2 g/d | — |
| Class or drug | 2019 guideline recommended dosing2 | Alternative dosing (CyDRi)20 |
|---|---|---|
| Glucocorticoids | Prednisolone or prednisone 1 mg/kg/d PO for a maximum of 4-6 weeks (followed by tapered withdrawal) | Dexamethasone 40 mg PO on days 1, 8, 15, and 22 |
| Cyclophosphamide | 1.5-2 mg/kg/d PO for a maximum of 6 weeks | 1000 mg IV on days 1 and 22 |
| Rituximab | 375 mg/m2 IV weekly for a maximum of 4 cycles | 100 mg IV on days 1, 8, 15, and 22 |
| Mycophenolate mofetil | 1 g/d for 1 week, followed by 2 g/d | — |
High-dose IV immunoglobulin and immune tolerance regimens with high-dose FVIII, which were developed for inhibitors in congenital hemophilia A, are generally not recommended in AHA.
Efficacy of IST and monitoring
Complete remission is achieved by 60% to 90% of patients according to observational studies (Table 2). Overall, the rate of achieving CR appeared lower with glucocorticoids alone compared to combination therapies. However, results must be interpreted with caution owing to the retrospective design of most studies. Time to achieve remission varied between a few weeks and many months. Throughout this period, the risk of bleeding remains high, as it has been observed that only achieving a FVIII level above 50% is fully protective.18
Efficacy of IST according to selected observational studies
| Study | N | Median age, y | Partial remission | Complete remission |
|---|---|---|---|---|
| EACH2 registry26 | 294 | 75 | NR | GC: 58% GC + Cy: 80% GC + Ri: 64% |
| GTH-AH 01/201012 | 102 | 74 | 83% | 61% |
| Spanish registry14 | 151 | 74 | NR | 84% |
| Dutch cohort study13 | 143 | 73 | NR | 79% |
| Chinese national registry27 | 187 | 52 | GC: 70% GC + Cy: 92% | GC: 49% GC + Cy: 83% |
| Budapest (CyDRi)20 | 32 | 77 | NR | 91% |
| Study | N | Median age, y | Partial remission | Complete remission |
|---|---|---|---|---|
| EACH2 registry26 | 294 | 75 | NR | GC: 58% GC + Cy: 80% GC + Ri: 64% |
| GTH-AH 01/201012 | 102 | 74 | 83% | 61% |
| Spanish registry14 | 151 | 74 | NR | 84% |
| Dutch cohort study13 | 143 | 73 | NR | 79% |
| Chinese national registry27 | 187 | 52 | GC: 70% GC + Cy: 92% | GC: 49% GC + Cy: 83% |
| Budapest (CyDRi)20 | 32 | 77 | NR | 91% |
Cy, cyclophosphamide; GC, glucocorticoid; NR, not reported; Ri, rituximab.
Close monitoring of FVIII levels is recommended until patients achieve CR, for 2 reasons: first, IST should be withdrawn or tapered as soon as PR is achieved; second, relapse occurs in approximately 20% of patients, most often early after achieving PR. After achieving CR, monthly monitoring is recommended during the first 6 months, every 2 to 3 months up to 12 months, and every 6 months during the second year and beyond, if possible.2
Monitoring of inhibitor levels can be helpful to guide IST. In particular, failure to decrease inhibitor levels during 2 to 4 weeks of first-line IST may guide the decision to use second-line options.
Risks of adverse events due to IST
Patient with AHA are often elderly and frail at the time of diagnosis. Glucocorticoids and other immunosuppressive drugs carry a high risk of adverse events, particularly in frail patients. The GTH-AH 01/2010 study followed a strictly prospective design and documented high rates of IST-related adverse events and mortality.12 Among the 34 deaths recorded in this study, the most common cause was infection (n = 16), followed by cardiovascular disorders (n = 6), underlying disorders (n = 3), and bleeding (n = 3). Fourteen deaths were directly attributed to IST. These findings indicate that mortality related to IST, especially infection, surpasses the current risk of fatal bleeding in AHA. Patients with a poor WHO performance status (>2) upon presentation had a 4-fold higher risk of mortality (Figure 1).
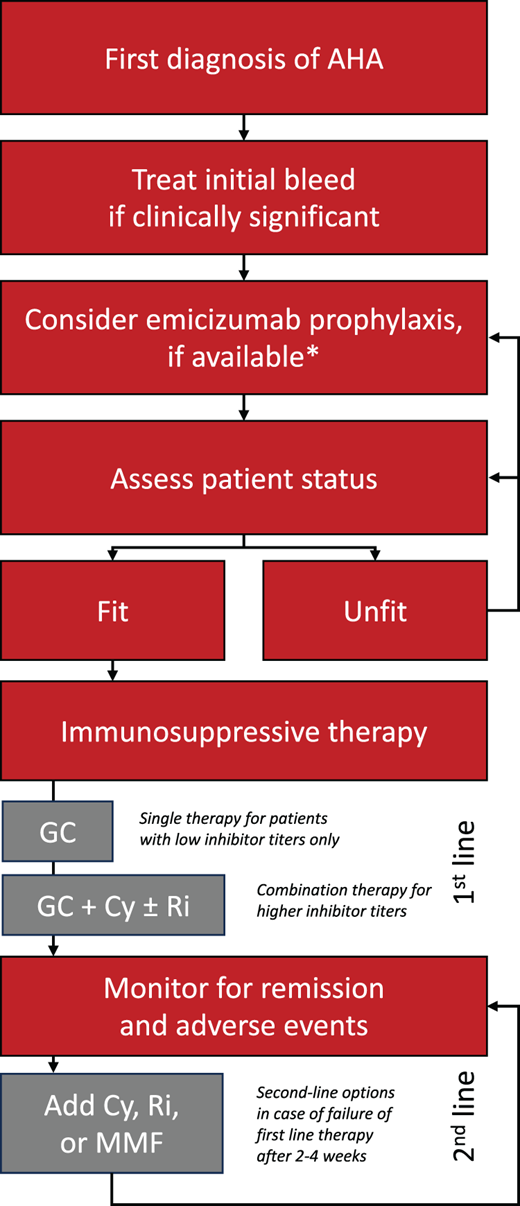
Suggested treatment algorithm. At the time of first diagnosis, most patients with AHA have active bleeding that requires hemostatic therapy. Once bleeding is under control, emicizumab prophylaxis should be considered to reduce the risk of bleed relapse and subsequent bleeding. Patients who are fit for IST should be offered first-line glucocorticoids (GCs), cyclophosphamide (Cy), and/or rituximab (Ri) (see text for details). Mycophenolate mofetil (MMF) is a second-line option. Patients who are not fit for IST should be reassessed in regular intervals and considered to be receive bleed prophylaxis with emicizumab. *Note: emicizumab is currently not licensed for AHA, except in Japan.
Suggested treatment algorithm. At the time of first diagnosis, most patients with AHA have active bleeding that requires hemostatic therapy. Once bleeding is under control, emicizumab prophylaxis should be considered to reduce the risk of bleed relapse and subsequent bleeding. Patients who are fit for IST should be offered first-line glucocorticoids (GCs), cyclophosphamide (Cy), and/or rituximab (Ri) (see text for details). Mycophenolate mofetil (MMF) is a second-line option. Patients who are not fit for IST should be reassessed in regular intervals and considered to be receive bleed prophylaxis with emicizumab. *Note: emicizumab is currently not licensed for AHA, except in Japan.
Therefore, careful consideration of the need for and contraindications to IST, as well as its intensity and timing, is crucial for frail patients with AHA. IST should be discontinued if severe side effects occur during treatment.
Data on antibiotic and antiviral prophylaxis are scarce, but the high risk of infection may justify its use in patients at risk. Cotrimoxazole (960 mg PO 3 times weekly) and low-dose acyclovir (400 mg PO once daily) might be reasonable options.
Prognostic factors for outcomes of IST
Inhibitor titer at baseline appears to be an important prognostic factor for achieving CR. This was already identified in a meta-analysis of 249 patients published in 20039 and also in a recently published registry from The Netherlands.13 In the latter study, patients with baseline inhibitor <20 BU/mL and mild bleeding at presentation had a 61% chance of CR with glucocorticoid therapy alone, compared to 7% of patients with >20 BU/mL and severe bleeding. Similar results were published from the GTH-AH 01/2010 study.12 Predictors for mortality were advanced age (>75 years), malignancy, and intensive care unit admission in the Dutch study.13 In the German- Austrian study, poor WHO performance status and malignancy predicted mortality.12
Newer IST regimens
The development of less toxic IST regimens remains a priority for research in IST. Mycophenolate mofetil was initiated either as first-line therapy in combination with glucocorticoids or as subsequent additional therapy in a single-center study of 11 patients.17 With the exception of 1 patient, who died from underlying lymphoma, all patients were alive and in PR at 1 year of follow-up. Two patients developed transient neutropenia, and infections occurred in 4 patients at 1 year.
Another single-center study of 25 patients with AHA used dexamethasone pulses of 40 mg (4 days in weeks 1, 2, 5, and 6) instead of daily prednisolone and stratified additional immunosuppressive drugs according to baseline characteristics.19 Results were similar to the GTH-AH 01/2010 study. Sixteen patients had grade 3 or 4 infection, and 2 patients died from infection.
A first-line combination therapy of glucocorticoids (dexamethasone 40 mg PO, days 1, 8, 15, and 22), cyclophosphamide (1000 mg IV, days 1 and 22), and rituximab (100 mg, days 1, 8, 15, and 22), designated as the CyDRi regimen, was recently reported from 2 centers in Budapest, Hungary.20 Of the 32 patients who were retrospectively identified and had received this regimen, 31 achieved CR. Infections were reported in 5 patients and treatment-related mortality in only 1 patient.
Special situations
Based on limited data, an approach similar to other patients was suggested in patients with pregnancy-associated AHA, although careful consideration may be required with the use of cytotoxic agents. The same applies to younger patients with AHA who are still in the reproductive age.
The usual principles of management, including IST, also apply to patients with malignancy-associated AHA.21 In patients with active solid tumors, IST will often be required to achieve remission before surgical resection or even biopsy of suspected lesions becomes feasible. About half of the malignancies associated with AHA are hematologic neoplasms.22 In these cases, treatment of the malignancy may in part overlap with IST (including glucocorticoids, rituximab, and cyclophosphamide), and the regimen should attempt to sufficiently treat both the underlying condition and AHA.
Patients with autoimmune disorders can develop AHA while already on immunosuppressive drugs, and combination therapies will often be required to induce remission. After achieving PR in these patients, IST would be reduced to doses of glucocorticoids or other immunosuppressants used before to control the underlying autoimmune disorder.
Future directions
Initial reports on the use of emicizumab in AHA are promising.23,24 Provided the efficacy and safety of emicizumab in patients with AHA are similar to patients with congenital hemophilia A, the drug reduces the requirement for early IST.25 Postponing IST until patients have recovered from sequelae of the initial bleeding symptoms, surgery, and frailty could reduce the risk of infectious complications and improve mortality. Likewise, emicizumab could reduce the risk of bleeding complications until remission of AHA is achieved. Prospective clinical trials are under way to address these questions.
Of similar importance is the development of safer IST protocols. Promising results from retrospective observations should be confirmed by prospective studies. These should also address the use of antibacterial and antiviral prophylaxis to reduce the risk of infection.
Conflict-of-interest disclosure
Andreas Tiede reports institutional grants for research and studies from Bayer, Biotest, Chugai, Novo Nordisk, Octapharma, Pfizer, Roche, SOBI, and Takeda and honoraria for lectures or consultancy from Bayer, Biomarin, Biotest, Chugai, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Roche, SOBI, and Takeda.
Off-label drug use
Andreas Tiede: emicizumab, rituximab, mycofenolate mofetil, cyclophosphamide.
