

Abstract
Acquired hemophilia A (AHA) is a rare disorder in which autoantibodies against factor VIII (FVIII) lead to a bleeding phenotype that varies from life-threatening to no bleeding at all. Prolonged activated partial thromboplastin times (APTT) in patients with a bleeding phenotype should be investigated to rule out AHA and should never be ignored in a preprocedure patient. Most inhibitors in AHA are heat and time dependent, so mixing studies performed only on an immediate mix are not useful: both lupus anticoagulants and treatment with direct oral anticoagulants can coexist with AHA and confound the diagnosis. Assays for intrinsic coagulation factors and von Willebrand factor should always be performed, regardless of the results of mixing studies. A Bethesda or modified Bethesda assay should be performed to quantify any inhibitor, and if susoctocog alfa (rpFVIII) is available, then an assay for cross-reacting antibodies should also be performed. At diagnosis and until complete remission, if the FVIII in the patient sample is >5 IU/dL, heat inactivation should be performed before the inhibitor assays are performed. While there are no conventional tests available to measure the effects of FVIII bypassing therapies, newer therapies may require monitoring, or their effects may need to be considered when choosing appropriate assays. Measurement of rpFVIII requires a 1-stage clotting assay, and measurement of patient FVIII while on emicizumab requires a chromogenic assay that does not contain human FX. Close communication is required between the treating clinicians and the laboratory to ensure that the correct tests are performed while patients are receiving treatments.
Learning Objectives
Understand the laboratory assays used for the differential diagnosis of acquired hemophilia A
Understand the assay considerations for laboratory monitoring of treatment and recovery in AHA with current and future treatment options
Laboratory diagnosis of acquired hemophilia A
Acquired hemophilia A (AHA), in which there are autoantibodies (inhibitors) against clotting factor VIII (FVIII), is a rare disorder that can occur in men or women.1 The bleeding phenotype varies from life-threatening to mild bleeding, and, in approximately 10% of presentations, no bleeding at all.2
In a patient with a bleeding phenotype, a prolonged activated partial thromboplastin time (APTT) should be investigated to rule out AHA, and in a preprocedure patient, APTT should never be ignored.2 Suspicion of AHA is increased in patients over the age of 60,3 during pregnancy, or for 1 year after giving birth.4
CLINICAL CASE (Presentation)
A 53-year-old female presented to the emergency department (ED) with a recent history of bruising but no symptoms of bleeding. Her APTT was 82 seconds (local reference range 21-31 seconds) with a normal prothrombin time and fibrinogen of 4.3 g/L. Liver function tests were normal. She had previously delivered 4 children by Caesarean section, and there was no known malignancy.
Mixing studies
Plasma mixing studies (MSs) are performed to aid in identifying the cause of a prolonged APTT, which can be due to the presence of a factor deficiency (eg, FVIII deficiency) or an inhibitor (eg, a lupus anticoagulant [LA]).5
The APTT performed on a 50:50 mix of patient's plasma with normal plasma and tested immediately was 33 seconds. There are up to 6 ways to define the presence of a correction in MS, with no consensus.5 The best method for an interpreting MS will depend on the normal plasma used and the factor-, LA- and anticoagulant-sensitivity of the reagent. Locally, a result of 33 seconds or below is corrected, and, therefore, the immediate MS in our patient corrected.
However, most inhibitors detected in AHA are heat and time dependent, and MSs require incubation for 2 hours.6 In AHA, MSs tested immediately are likely to be similar to those seen in patients with a single factor deficiency and no inhibitor.5 In our patient, the APTT in the incubated MS was 75 seconds, confirming the presence of a time-dependent inhibitor.
Of the last 50 AHA patients presenting at our center, 9 (18%) had immediate-acting inhibitors, which have the potential to bind to the FVIII in the deficient plasmas used in 1-stage clotting factor assays (OSCAs), prolonging clotting times and showing an apparent reduction in factors IX (FIX), XI (FXI), and/or XII (FXII).7 These interferences can be identified by using multiple samples dilutions8 and by repeating the OSCA using higher dilutions an accurate result may be determined.
Patients presenting with AHA may also be taking direct oral anticoagulants. In particular, the presence of oral direct FXa inhibitors (DFXaIs) may be difficult to ascertain by screening tests alone, and MSs in the presence of DFXaIs are likely to show noncorrection in both immediate and incubated mixing studies. A suitably calibrated assay to detect the presence of DFXaIs may be useful.
Factor assays
Assays for FVIII, FIX, FXI, and von Willebrand factor (VWF) (activity and antigen)10 must be performed at presentation, regardless of the results of mixing studies,9 as it cannot be assumed that AHA is the only possible diagnosis. Occasionally, patients with mild hemophilia A or B, mild von Willebrand disease (VWD), or deficiencies of FXI may reach old age without being diagnosed or having an APTT measured. Acquired von Willebrand syndrome and acquired FXI deficiency may also be encountered. An FVIII to VWF antigen ratio may be useful in differentiating type 1 VWD from AHA, mild hemophilia A, and type 2N VWD. Although not associated with a bleeding phenotype, deficiency of FXII is a frequent cause of a prolonged APTT, so an FXII assay may be included in the initial investigations.
In our patient, FVIII was <1 IU/dL by both an OSCA and a chromogenic substrate assay (CSA); the VWF:GpIbM activity assay was 160 IU/dL, and VWF:Ag was 220 IU/dL. Factors IX, XI and XII were normal.
Of the last 50 AHA patients presenting at our center, the median FVIII level using a CSA was 2 IU/dL (range <1 to 40 IU/dL): 13 (26%) had FVIII <1 IU/dL, and 12 (24%) had FVIII between 10 and 25 IU/dL, similar to the observations of the European Acquired Haemophilia Registry.3 Only 2 of these 50 patients had discrepancies between their OSCA and CSA (13 and 2 IU/dL; 8 and 22 IU/dL); the choice of assay did not make any difference to the AHA diagnosis. Laboratories using LA-sensitive reagents in OSCA may see falsely reduced FVIII levels;2 these laboratories should confirm all reduced FVIII in OSCAs by also using a CSA, as these assays are usually insensitive to LA.11,12
The presence of DFXaIs may cause FVIII to appear reduced on a CSA, and, depending on the reagent and type and concentration of DFXaI, the OSCA may also be affected. Along with a prolonged APTT and noncorrection in the MS, there is potential for the misdiagnosis of AHA in these patients. The use of an activated carbon product to remove DFXaIs from the sample in vitro could be considered; although the effect of these products on OSCAs and CSAs for FVIII has been described,13 there are limited data on the use of these assays in patients with AHA. In our patient, there was no history of DFXaI use.
Assays for LA using APTT are difficult to interpret in AHA, but dilute Russell viper venom time results are not affected. The coexistence of an antiphospholipid antibody is not unusual in AHA, as both are autoimmune disorders that may coexist in the same patient. In our patient, the dilute Russell's viper venom time was normal.
All OSCA factor assays should be performed at multiple dilutions.8 This will allow the accurate quantitation of factor activities in FIX, FXI, and FXII assays in the presence of very strong or immediate-acting inhibitors to FVIII, in the presence of high doses of DFXaIs, or in the presence of a strong LA.
Inhibitor assays
If a reduced FVIII (<50 IU/dL) is detected,3,14 a standard Bethesda15 or Nijmegen-modified Bethesda16 should be performed. Patient plasma is incubated with normal plasma15 or buffered normal plasma16 as a source of FVIII, and the amount of FVIII measurable in the mixture after 2 hours is compared to a negative control (inhibitor-free FVIII-deficient plasma incubated with normal plasma). This residual FVIII is expressed as a percentage ([patient ÷ control] × 100). Samples should be titrated in FVIII-deficient plasma until the inhibitor is not detectable in the dilution tested.
If the amount of FVIII in the patient sample is above 5 IU/dL, incubating the sample for 30 minutes at 56°C will denature all the endogenous FVIII present in the sample and leave the inhibitor suitable for use in the Bethesda assay,17 improving assay sensitivity.18
In patients with congenital hemophilia A, alloantibodies to FVIII show linear type 1 kinetics in the Bethesda assay, whereas the autoantibodies in AHA often display complex, nonlinear type 2 kinetics.19 The results for our patient with type 2 kinetics are shown in Table 1 and Figure 1.

Residual FVIII against a sample dilution for a type 1 inhibitor often seen in congenital hemophilia (pink line) and against the type 2 inhibitor seen in a clinical case (black line). The blue shaded area denotes the area between 30% and 70% residual FVIII, where the Bethesda assay is considered to be accurate. For type 1 inhibitors, only 1 or 2 dilutions will give interpretable results (and the same calculated titer), whereas for type 2 inhibitors, multiple dilutions will give interpretable results, and the calculated titer will be different for each one.
Residual FVIII against a sample dilution for a type 1 inhibitor often seen in congenital hemophilia (pink line) and against the type 2 inhibitor seen in a clinical case (black line). The blue shaded area denotes the area between 30% and 70% residual FVIII, where the Bethesda assay is considered to be accurate. For type 1 inhibitors, only 1 or 2 dilutions will give interpretable results (and the same calculated titer), whereas for type 2 inhibitors, multiple dilutions will give interpretable results, and the calculated titer will be different for each one.
Bethesda assay results in patient with AHA
| Sample dilution | Residual FVIII (%) | Inhibitor titer in this dilution | Inhibitor titer (BU/mL) |
|---|---|---|---|
| Neat | 27.6 | >2.00 | >2.0 |
| 2 | 38.5 | 1.38 | [1.38 × 2 = ] 2.8 |
| 4 | 45.4 | 1.14 | [1.14 × 4 = ] 4.6 |
| 8 | 50.3 | 0.99 | [0.99 × 8 = ] 7.9 |
| 16 | 57.7 | 0.79 | [0.79 × 16 = ] 12.6 |
| 32 | 71.6 | <0.50 | <16.0 |
| Sample dilution | Residual FVIII (%) | Inhibitor titer in this dilution | Inhibitor titer (BU/mL) |
|---|---|---|---|
| Neat | 27.6 | >2.00 | >2.0 |
| 2 | 38.5 | 1.38 | [1.38 × 2 = ] 2.8 |
| 4 | 45.4 | 1.14 | [1.14 × 4 = ] 4.6 |
| 8 | 50.3 | 0.99 | [0.99 × 8 = ] 7.9 |
| 16 | 57.7 | 0.79 | [0.79 × 16 = ] 12.6 |
| 32 | 71.6 | <0.50 | <16.0 |
The type 2 kinetics of the autoantibody gives the appearance that the inhibitor gets stronger the more it is diluted. For consistency, the dilution that gives closest to 50% residual FVIII (in italics) is used to determine the inhibitor titer that is reported to clinicians—in this case, 7.9 BU/mL.
Some low-titer inhibitors with type 2 kinetics may not be detectable in neat plasma but may be detectable in the next titration. Therefore, it may be advisable to test neat plasma and plasma diluted by at least ½ in all cases.
Measurement of residual FVIII after the incubation step in the Bethesda assay can be by OSCA or CSA. The CSA is less likely to be susceptible to interference from an LA than the OSCA if the local APTT reagent in use is LA-sensitive;20 both assays are susceptible to interference from direct oral anticoagulants. There is a significant risk that if the presence of a DFXaI is not detected, a patient could have a prolonged APTT, noncorrection in the MSs, a reduced FVIII, and a positive Bethesda assay, resulting in an erroneous diagnosis of AHA.
Our patient had an inhibitor that was quantitated at 7.9 BU/mL at presentation with type 2 kinetics.
Of the last 50 patients diagnosed with AHA at our center, the median inhibitor titer on presentation was 9.5 BU/mL (range 1.4 to 2224 BU/mL): 10 had type 1 kinetics and 40 had type 2 kinetics. Recent international guidelines2 suggest that those with FVIII <1 IU/dL or an inhibitor titer >20 BU/mL are less likely to achieve partial remission in the first 21 days if treated with corticosteroids alone and should therefore receive combined immunotherapy.
Enzyme-linked immunosorbent assay
Commercially-available anti-FVIII enzyme-linked immunosorbent assay (ELISA) has been shown to be sensitive and specific for diagnosing AHA and may be used as an adjunct or alternative to a Bethesda assay,2,21 particularly if there is suspicion of LA or DFXaI interference in the Bethesda assay. Native or heat-inactivated plasma or serum can be used.
It should be noted that positive anti-FVIII results can be found with ELISA in a significant number of individuals who do not have congenital or acquired hemophilila A.22
Porcine inhibitor assays
Patients with AHA may be given recombinant porcine FVIII, susoctocog alfa (rpFVIII), to treat bleeds or to cover minor or major surgery. If rpFVIII is an available treatment option, newly diagnosed AHA patients should have another inhibitor assay performed on the same sample, using rpFVIII as the source of FVIII in the Bethesda assay.
In studies, cross-reacting anti-rpFVIII antibodies were detectable in 44%23 and 49%24 of newly presenting patients with AHA. A patient may be suitable to receive rpFVIII to treat bleeds or to cover minor or major surgery if there is no anti-rpFVIII inhibitor detectable. Patients with anti-rpFVIII inhibitors below 20 BU/mL can receive rpFVIII but are likely to require more to maintain trough levels above 50 IU/dL.25
Anti-rpFVIII inhibitors may exhibit type 1 or type 2 kinetics. An OSCA method is preferred for the measurement of residual rpFVIII in the Bethesda assay.26
When our patient presented, rpFVIII was not licensed; when the drug became available, her anti–human FVIII inhibitor level was 1.3 BU/mL, but no anti-rpFVIII inhibitor was detectable.
A suggested testing algorithm for patients suspected of AHA is shown in Figure 2.
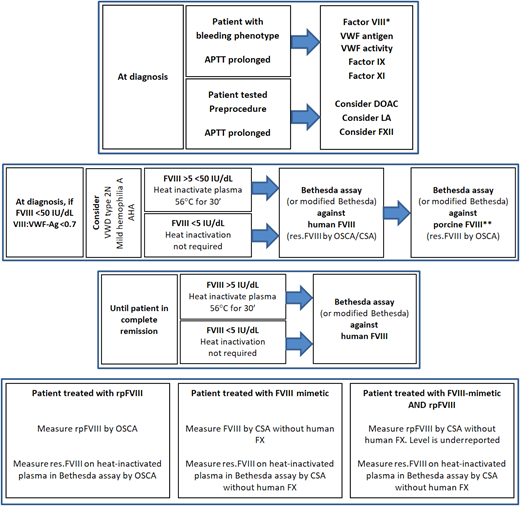
Algorithm for Acquired Haemophilia A diagnosis and monitoring. *One-stage clotting assay and/or chromogenic substrate assay. **Only required if treatment with susoctocog alfa is available locally. AHA, acquired haemophilia A; APTT, activated partial thromboplastin time; CSA, chromogenic substrate assay; DOAC, direct oral anticoagulant; FVIII, factor VIII; FX, factor X; LA, lupus anticoagulant; OSCA, one-stage clotting assay; res.FVIII, residual FVIII; rpFVIII, susoctocog alfa; VII:VWF-Ag, factor VIII to von Willebrand factor antigen ratio; VWF, von Willebrand factor.
Algorithm for Acquired Haemophilia A diagnosis and monitoring. *One-stage clotting assay and/or chromogenic substrate assay. **Only required if treatment with susoctocog alfa is available locally. AHA, acquired haemophilia A; APTT, activated partial thromboplastin time; CSA, chromogenic substrate assay; DOAC, direct oral anticoagulant; FVIII, factor VIII; FX, factor X; LA, lupus anticoagulant; OSCA, one-stage clotting assay; res.FVIII, residual FVIII; rpFVIII, susoctocog alfa; VII:VWF-Ag, factor VIII to von Willebrand factor antigen ratio; VWF, von Willebrand factor.
CLINICAL CASE (Monitoring)
Monitoring of patients on bypassing agents
No specific tests exist for monitoring treatment with activated prothrombin complex concentrates or recombinant activated factor VII. Thrombin generation assays can be used, but despite attempts to standardize testing, results in patients with hemophilia may not be comparable between those with the same FVIII activity;27 results in an individual may show improvement with therapy, but there are no therapeutic targets for thrombin generation assay parameters.
Remission/relapse
Partial remission has been defined as FVIII >50 IU/dL and no bleeding after stopping any hemostatic treatment for at least 24 hours.14 Complete remission is defined as normal FVIII with no detectable inhibitor and either (1) immunosuppression either stopped or reduced to levels used before AHA diagnosis or (2) prednisolone <15 mg/d and all other immunosuppression stopped.14 Neither of these definitions specifies whether the inhibitor should be measured by a Bethesda, Nijmegen-modified, or ELISA method, nor whether heat-inactivated samples should be used. Inhibitors often persist in patients with AHA even when FVIII levels have reached normal levels,18,28 so local practice is to use heat inactivation in a Nijmegen-modified assay.
Laboratories should continue to monitor FVIII levels and test for the presence of an inhibitor regardless of FVIII level until immunosuppression is stopped.
Repeated measure of inhibitors during recovery
In our patient, FVIII levels exceeded 50 IU/dL in less than 4 weeks (month 1), and no inhibitor was detected (Figure 3). At that time, heat inactivation of samples was not performed, making inhibitors difficult to detect in a Bethesda assay when FVIII levels exceed ∼20 IU/dL.
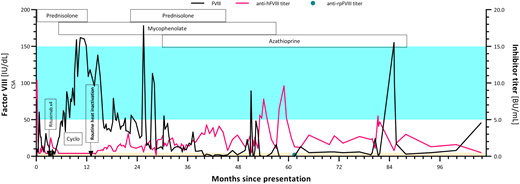
FVIII activity (left axis) and inhibitor titer (right axis) from diagnosis to first partial remission. Shaded blue area represents the reference range for factor VIII activity; shaded orange area represents the reference range for the Bethesda assay. anti-hFVIII, antihuman factor VIII; anti-rpFVIII, antirecombinant porcine factor VIII; CSA, chromogenic substrate assay; Cyclo, cyclophosphamide; FVIII, factor VIII.
FVIII activity (left axis) and inhibitor titer (right axis) from diagnosis to first partial remission. Shaded blue area represents the reference range for factor VIII activity; shaded orange area represents the reference range for the Bethesda assay. anti-hFVIII, antihuman factor VIII; anti-rpFVIII, antirecombinant porcine factor VIII; CSA, chromogenic substrate assay; Cyclo, cyclophosphamide; FVIII, factor VIII.
A prednisolone wean was commenced, but FVIII levels immediately dropped. Immunosuppression continued, and brief recoveries in FVIII and reductions in inhibitor titer (without heat-inactivation of plasma) were seen 7 to 14 months after the patient first presented (Figure 3).
Heat inactivation of samples became routine practice in this laboratory around 13 months after our patient first presented (Figure 3): this coincided with a drop in FVIII and a low-titer inhibitor being detectable from that point onwards.
Over the course of the next 5½ years (18 months onwards), our patient continued to receive immunosuppression, with FVIII occasionally peeking into the reference range, but her inhibitor remained detectable throughout (Figure 3) and complete remission was never achieved.
CLINICAL CASE (Undergoing surgery)
Nine years after diagnosis, our patient presented to the ED with abdominal pain secondary to a benign ovarian cyst. During an ED admission 4 weeks later, clinicians found that the cyst had doubled in size and, despite a normal CA125 level, it appeared to be malignant on computed tomography scanning. Peritoneal metastases were also identified, and she required surgery. Immunosuppression with prednisolone was reinitiated 2 weeks before a laparotomy, and FVIII levels reached 221 IU/dL by the day of surgery. No additional products were required for the surgery nor for when she required repair of a colonic anastomosis 7 days later. Stage 3C ovarian carcinoma was diagnosed with metastases in the bowel and bladder, which were removed. She had an episode of septic shock 2 weeks after surgery when her FVIII levels reached 436 IU/dL (Figure 4).
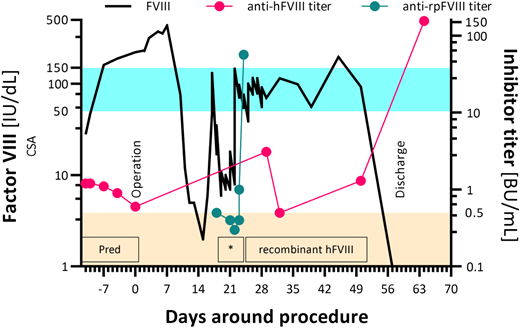
Factor VIII activity (left axis) and inhibitor titer (right axis) during hospital stay, 10 years after diagnosis: note logarithmic scale on y-axes. Shaded blue area represents the reference range for factor VIII activity; shaded orange area represents the reference range for the Bethesda assay. *Denotes the time when the patient received susoctocog alfa. anti-hFVIII, anti-human factor VIII; anti-rpFVIII, anti–recombinant porcine factor VIII; CSA, chromogenic substrate assay; FVIII, factor VIII; Pred, prednisolone.
Factor VIII activity (left axis) and inhibitor titer (right axis) during hospital stay, 10 years after diagnosis: note logarithmic scale on y-axes. Shaded blue area represents the reference range for factor VIII activity; shaded orange area represents the reference range for the Bethesda assay. *Denotes the time when the patient received susoctocog alfa. anti-hFVIII, anti-human factor VIII; anti-rpFVIII, anti–recombinant porcine factor VIII; CSA, chromogenic substrate assay; FVIII, factor VIII; Pred, prednisolone.
Treatment of patients with susocotocog alfa
Patients with AHA who are given rpFVIII to treat bleeds or to cover minor or major surgery are given a standard dose of 200 U/kg followed by repeat doses to maintain levels in the target range,2 although there is evidence that a dose of 100 U/kg may be sufficient.29,30 Frequent monitoring of rpFVIII levels at trough and peak is required.31,32
The OSCA should be used to measure rpFVIII,26 and VWF level in the FVIII-deficient plasma used may also be required.33 In a field study,34 the mean recovery for rpFVIII by CSA was 53% (range 44%-61%), and the mean ratio of OSCA:CSA was 1.85, with interlaboratory variation being higher. Recovery of rpFVIII should be measured 2 to 3 times per week to evaluate for the development of inhibitors against rpFVIII26 ; if recovery of rpFVIII is less than expected, retesting for antihuman and anti-rpFVIII may be considered. When rpFVIII was assessed in a prospective, single-arm clinical study, 5 out of 18 (28%) patients who did not have detectable anti-rpFVIII inhibitors at baseline developed anti-rpFVIII antibodies after 8-85 days.25
In our patient, FVIII levels dropped 3 weeks after surgery, and rpFVIII prophylaxis was started. Immediately before prophylaxis, her anti-rpFVIII inhibitor result was negative, but a week later, the anti-rpFVIII titer jumped to 56.8 BU/mL; prophylaxis was switched to recombinant human FVIII. A week later, her FVIII became undetectable, and her antihuman FVIII inhibitor peaked at 155.2 BU/mL (Figure 4). However, she was successfully discharged home, and chemotherapy was planned.
Three weeks following discharge she was found to have liver and lymph node metastases, after which she was palliated. She died 5 weeks later.
Other treatments
Treatment of patients with factor VIII mimetics
Emicizumab is a recombinant bispecific monoclonal antibody with FVIII-mimetic activity that is licensed for use in patients with congenital hemophilia A with or without inhibitors. In some case reports and case series, emicizumab has been used off-label in patients with AHA.35 A prospective multicenter phase 3 study of emicizumab prophylaxis in AHA has recently commenced, and it is likely that this drug will be used to prevent bleeds in AHA.36
Emicizumab does not require monitoring, but FVIII levels may need to be measured in AHA to assess if patients are in partial or complete remission. Emicizumab interferes with APTT-based OSCAs and also with CSAs that use human FX.37,38 Therefore, CSAs that use bovine FX must be used to calculate both the patient's own FVIII and the residual FVIII in any Bethesda inhibitor assay.39
Heat inactivation up to 58°C does not denature emicizumab.40
Patients on emicizumab who bleed and are given rpFVIII
Emicizumab-calibrated modified OSCAs and CSAs that include human FX in the reagents are known to lead to overestimation of FVIII of up to 89% when compared to recombinant human FVIII,41 and it is highly likely that this will also apply to assays of rpFVIII in the presence of emicizumab. Therefore, although the preferred assay for rpFVIII is an OSCA, the only assays that available to measure rpFVIII levels in these patients will be CSAs that do not contain human FX reagents.26 When monitoring such patients, it will be important to remember that CSA underestimates rpFVIII by 44%-61%.34
Other FVIII-mimetics may become available in the future: consideration of their mode of action will be needed when selecting appropriate assays.
Conclusion
Acquired hemophilia A is a rare disease that sometimes relies on a laboratory diagnosis to be correctly made, especially in patients with a mild or absent bleeding phenotype at presentation. This includes a full investigation of a prolonged APTT regardless of the results of mixing studies, including assessing for FVIII, FIX, FXI, FXII, VWF activity, and VWF antigen, with consideration being given to the coexisting presence of oral anticoagulants and/or LA.
An inhibitor assay using a Bethesda or modified Bethesda assay should be performed using human plasma FVIII as the source of FVIII: residual FVIII can be measured using an OSCA or a CSA, although a CSA may be more specific. If rpFVIII is a potential treatment option, it should be used as the source of FVIII in an additional inhibitor assay, and an OSCA should be used to measure residual FVIII. An ELISA for anti-FVIII antibodies can be performed if it is not possible to exclude the presence of oral anticoagulants or LA.
Careful choice of laboratory assays is required for newer therapies for AHA, as measurement of rpFVIII requires an OSCA assay, and measurement of patient FVIII (or residual FVIII in a Bethesda or modified Bethesda assay) while on factor VIII mimetics requires a CSA that does not contain human FX. Compromises need to be made if patients receive rpFVIII to treat bleeds while on factor VIII mimetics, and clinicians and laboratories need to work closely together to ensure that the correct assays are performed and interpreted correctly.
Acknowledgments
Sean Platton would like to acknowledge Minal Dave from Barts Health NHS Trust and Annette Bowyer from the Sheffield Teaching Hospitals NHS Foundation Trust for their help in proofreading this manuscript.
Conflict-of-interest disclosure
Sean Platton has received honoraria from Novo Nordisk, Pfizer, and Sanofi and travel support from Sysmex UK.
Suthesh Sivapalaratnam has received consulting fees from HemAb.
Priyanka Raheja has received grant/travel support from CSL Behring, Sobi, and Takeda and honoraria from Idogen, Sigilon, Sobi, and Pfizer.
Off-label drug use
Sean Platton: the use of emicizumab for patients with AHA is off-label.
Suthesh Sivapalaratnam: the use of emicizumab for patients with AHA is off-label.
Priyanka Raheja: the use of emicizumab for patients with AHA is off-label.
