

Abstract
Novel therapies in multiple myeloma (MM) have increased the rates of conventional complete remission (CR) in patients. However, patients in CR can have highly heterogeneous outcomes. Novel and more sensitive methods of assessing residual disease burden after therapy will help prognosticate this group better and, ideally, allow individualized therapy adjustments based on response depth in the future. Here, we review novel bone marrow, peripheral blood, and imaging methods for assessing myeloma burden and discuss the opportunities and limitations of incorporating these in everyday clinical practice.
Learning Objectives
Understand the utility of bone marrow minimal residual disease testing in clinical practice
Understand the role of PET/CT and MRI in assessing response to therapy and prognosis in MM
Outline the clinical use of mass spectrometry–based methods for assessing monoclonal proteins in MM
CLINICAL CASE 1
A 65-year-old woman presents with newly diagnosed immunoglobulin G kappa (IgG kappa) multiple myeloma (MM) with high-risk (HR) features (del17p, t(4;14) and Revised International Scoring System [R-ISS] stage III). Positron emission tomography/computed tomography (PET/CT) demonstrates soft tissue plasmacytomas. She receives induction therapy with lenalidomide, bortezomib, and dexamethasone (VRd) followed by myeloablative doses of melphalan and autologous stem cell transplant (ASCT). Pre-ASCT testing reveals she has achieved a minimal residual disease (MRD)-negative complete remission (CR) measured at a 10−5 sensitivity threshold using flow cytometry, which is maintained at day 100 plus post ASCT. She elects to have maintenance therapy with lenalidomide and bortezomib. She wants to know what an MRD-negative CR means for the outcome and management of her HR MM.
CLINICAL CASE 2
An 80-year-old man presents with newly diagnosed IgG kappa MM with no HR features and normal levels of serum albumin, lactate dehydrogenase, and beta-2-microglobulin (R-ISS stage I). He receives therapy with daratumumab, lenalidomide, and dexamethasone. After 6 months his immunofixation (IFE) remains positive for a circulating IgG kappa monoclonal protein in the blood, and a bone marrow (BM) biopsy is performed, which is MRD positive when measured at a 10−5 sensitivity threshold using flow cytometry. He wants to know what this means for the outcome and management of his MM.
How, why, and when should BM-based MRD testing be performed in patients with MM?
Multidrug combinations of novel agents can achieve conventional CR (defined as negative IFE on the serum and urine, the disappearance of any soft tissue plasmacytomas, and less than 5% plasma cells in BM aspirates) in more than two-thirds of patients.1-3 Trial and “real-world” data consistently demonstrate that both transplant eligible and ineligible patients achieving CR after first-line therapy tend to live longer.1-5 Since the outcomes of patients in conventional CR are variable, more sensitive measurements of residual disease are required to better prognosticate these patients, with the goal of adjusting therapies to improve patients' outcomes and lives. MRD refers to residual disease in the BM space that is below the limit of detection of conventional tests. However, when the term “MRD” is used in the literature, it frequently does not differentiate between MRD-negative CR (no circulating monoclonal protein) and MRD negativity in the BM with a circulating monoclonal protein (at best, classified as a very good partial remission). This distinction can frequently complicate the interpretation of MRD data.
Currently, next generation flow cytometry (NGF) and next generation sequencing (NGS) are the 2 methods used to detect MRD in the BM. The main differences between them are shown in Table 1. These methods have been reviewed extensively.6,7 Briefly, NGF requires a slightly larger initial sample to be processed within 48 hours (~10 million cells vs ~1 million for NGS, which translates to usually no more than 1 ml of additional BM aspirate). It is also slightly less sensitive than NGS (10−5 vs 10−6). Its performance compared to NGS in patients treated with CD38 antibodies is not well established. However, it is faster and does not require a baseline sample for clone identification. Reassuringly, the concordance between NGS and the EuroFlow NGF method is high,8,9 although this may not be true for other NGF methods.10 The prognostic significance of MRD negativity has been demonstrated in several trials and trial-level metanalyses at various time points across the disease spectrum, regardless of disease or patient characteristics.7,11 A recent metanalysis of 93 publications from 45 studies demonstrated that MRD negativity is associated with improved progression-free (PFS) and overall survival (OS) in transplant-eligible or transplant-ineligible patients at diagnosis or relapse.12 Increasing MRD sensitivity thresholds showed a trend toward improved PFS and OS, and a threshold of 10−5 or 10−6 is currently considered standard. However, these studies did not always differentiate between MRD-negative CR and MRD-negative non-CR. Reassuringly, MRD negativity remained prognostic in both CR and very good partial remission patients. Additionally, MRD negativity was associated with a similar PFS and OS benefit in HR and non-HR patients. It is worth noting though, that achieving MRD negativity in HR patients in real-world retrospective studies did not always translate into superior PFS/OS.13
MRD methods used for BM assessment
| NGF | NGS | |
|---|---|---|
| Applicability | Nearly 100% | ≥90% (limited mainly by somatic hypermutation of the originally identified malignant clone) |
| Baseline sample | Not required | Required; patient-specific probes are not required |
| Quantity of sample required | Up to 10 million for 10−6 sensitivity | 2-3 million for 10−6 sensitivity |
| Sample processing | Needs to be processed within 48 h. MRD testing is frequently “reflexed” in patients with no BM involvement despite residual detectable M protein while awaiting M protein quantification tests. | Can be delayed; can use both fresh and stored samples |
| Sample quality control | Yes (highly reproducible detection of hemodilution in each sample) | No |
| Sensitivity | ≥1 in10−5 | ≥1 in 10−6 (only limited by the number of cells provided by the biopsy) |
| Additional information | Ability to evaluate BM microenvironment and hematologic subpopulations | Information about immunoglobulin gene repertoire of B cells in the studied patient samples |
| Turnaround for results | A few hours with automated software available | Approximately 1 wk |
| Availability | Eight or more color flow cytometry requires more experienced laboratories. Many laboratories have adopted the EuroFlow laboratory protocols and use the EuroFlow MRD tubes. | So far limited to 1 company/platform that has US Food and Drug Administration approval |
| Cost | Approximately US $500/sample | Approximately US $1100 |
| NGF | NGS | |
|---|---|---|
| Applicability | Nearly 100% | ≥90% (limited mainly by somatic hypermutation of the originally identified malignant clone) |
| Baseline sample | Not required | Required; patient-specific probes are not required |
| Quantity of sample required | Up to 10 million for 10−6 sensitivity | 2-3 million for 10−6 sensitivity |
| Sample processing | Needs to be processed within 48 h. MRD testing is frequently “reflexed” in patients with no BM involvement despite residual detectable M protein while awaiting M protein quantification tests. | Can be delayed; can use both fresh and stored samples |
| Sample quality control | Yes (highly reproducible detection of hemodilution in each sample) | No |
| Sensitivity | ≥1 in10−5 | ≥1 in 10−6 (only limited by the number of cells provided by the biopsy) |
| Additional information | Ability to evaluate BM microenvironment and hematologic subpopulations | Information about immunoglobulin gene repertoire of B cells in the studied patient samples |
| Turnaround for results | A few hours with automated software available | Approximately 1 wk |
| Availability | Eight or more color flow cytometry requires more experienced laboratories. Many laboratories have adopted the EuroFlow laboratory protocols and use the EuroFlow MRD tubes. | So far limited to 1 company/platform that has US Food and Drug Administration approval |
| Cost | Approximately US $500/sample | Approximately US $1100 |
It is unclear if achieving MRD negativity in newly diagnosed patients abrogates the negative prognostic impact of HR cytogenetics. HR patients can achieve MRD negativity at similar rates as non-HR patients, although this may not be true for 17p deletion.14 The PETHEMA/GEM2012MENOS65 trial compared 2 different conditioning regimens in patients receiving VRd induction.15 In this trial, the PFS of MRD-negative patients after the end of consolidation was similar in R-ISS stage III and R-ISS stage I or II disease.15 In the PETHEMA/GEM2010MAS65 trial,16 which randomized transplant-ineligible patients to either melphalan, bortezomib, and prednisone and lenalidomide-dexamethasone (Rd) maintenance or alternating melphalan, bortezomib, and prednisone and Rd, the time to progression for MRD-negative patients, both HR and non-HR, was the same. However, MRD analyses of the IFM 2009 trial, comparing early vs late ASCT in patients treated with VRd induction, showed that the PFS of MRD- negative HR patients was worse than that of non-HR patients if MRD was assessed at the start of maintenance therapy, although it was no different if assessed at the end of maintenance therapy, underlying the importance of sustained MRD negativity in HR patients.14
MRD-negative HR patients also had worse outcomes compared to MRD-negative non-HR patients in the Myeloma XI trial,3 which compared response-adapted intensive vs nonintensive treatment strategies during induction, consolidation, and maintenance. Finally, unpublished analyses of the CASSIOPEIA trial suggest that MRD-negative HR patients do not have improved outcomes compared to MRD-positive patients.17 These data are limited by the small number of HR patients included in the subgroup analyses.
The treatment strategy of achieving MRD negativity may not be important. This was demonstrated in an analysis of the CASSIOPEIA trial,18 which evaluated the addition of daratumumab to a triplet induction regimen and found that MRD-negative patients had similar PFS irrespective of the treatment arm to which they were randomized. However, in IFM2009 PFS was improved in MRD-negative patients randomized to receive up-front transplant when MRD was assessed prior to the initiation of maintenance, although PFS was no different when MRD was assessed after 12 months of maintenance.14
We can therefore inform both patients in the clinical cases that achieving an MRD-negative CR after initial therapy is associated with longer remission and survival. For patient 1, it likely does not abrogate the negative impact of HR cytogenetics. For both patients, there is no evidence to support modifying therapy based on MRD results.
CLINICAL CASE 1 AND 2 (Continued)
Nine months later patient 1 becomes MRD positive but remains in hematologic CR. In patient 2, a repeat BM biopsy 12 months after the initiation of therapy is now consistent with MRD negativity. They want to know what this means.
What is the significance of sustained MRD negativity or late conversion to MRD negativity?
Response kinetics need to be considered in relation to MRD testing. The Myeloma XI trial showed that MRD-positive patients at 3 months or more post ASCT converting to MRD-negative at 9 months or more had similarly favorable outcomes to patients achieving MRD-negative disease for 3 months or more and maintaining it for at least 9 months, and vice versa. Short-lived MRD negativity (negative at 3 months but positive at 9 months) had similar outcomes as MRD positivity at 3 or more months.3 Additionally, sustained MRD negativity is usually tested at 6- and 12-month intervals and has been consistently associated with improvements in PFS in both transplant-eligible and transplant-ineligible newly diagnosed or relapsed patients.3,19,20 Conversion to MRD positive from MRD negative even without any other evidence of disease progression is associated with a worse prognosis. A recent large single-center analysis showed that conversion to MRD positive was associated with clinical relapse in 72% of patients at a median of 12 months and a biochemical-only relapse at a median of 6 months.21 Additionally, several studies suggest that patients with gradual hematologic responses have better outcomes compared to patients achieving early deep responses,22,23 reflecting perhaps a more indolent disease biology. Early attainment of MRD negativity was associated with a shorter time to MRD-positive conversion,21 underlying the importance of serial MRD assessments, especially for HR patients.
We can therefore inform patient 1 that conversion to MRD positivity is a bad prognostic sign. While there are no data to inform changes in therapy in MRD-positive conversion, many experts would recommend changing therapy in this fit HR patient given the extremely poor outcomes of this group. We can inform patient 2 that conversion to an MRD-negative state is a favorable prognostic sign.
CLINICAL CASE 1 (Continued)
The patient is disappointed and asks if there is a role for a PET scan or magnetic resonance imaging (MRI) to detect early signs of bone damage.
What is the role of advanced imaging in assessing MM burden?
BM assessment has limitations. BM involvement by MM can be “patchy” and disease “hot spots” can be missed after a single biopsy attempt. MM can be predominantly extramedullary (EM), and finally, BM biopsies are not convenient. Whole-body MRI or PET/CT can provide additional prognostic information in patients achieving deep hematologic responses and, depending on the findings, may lead to a change in therapy if significant MM burden is identified.24
The association of a positive PET/CT with MRD status and its prognostic significance have been evaluated in several studies. Patients with MRD-negative BM can have PET avid lesions in 12% to 20% of cases after first-line therapy and up to 50% in relapsed disease, consistent with high rates of oligosecretory EM disease in this setting.25,26 In the PETHEMA/GEM2012MENOS65 trial,15 7 of the 14 MRD-negative patients relapsing in the study remained BM MRD negative upon relapse and had evidence of plasmacytomas, which underlines the importance of EM disease assessment. PET-positive patients consistently have worse outcomes regardless of MRD or hematologic response status.24-27 The impact of MRI findings on prognosis is less clear. MRI negativity in patients early after therapy in the IFM 2009 trial was not associated with PFS or OS.26 This is likely because of the lower sensitivity of MRI in differentiating active vs old MM lesions and therefore the smaller number of MRI-negative patients. Novel MRI techniques may be superior in differentiating between active and old MM lesions, but more studies are needed to establish their value for this. In our practice, PET/CT is used more frequently for the evaluation of residual bone disease after therapy, and the results are considered in conjunction with the patient's hematologic response and clinical picture.
In this case performing a PET/CT scan is reasonable, especially considering the EM disease at diagnosis, her early conversion to MRD positivity, and her HR status.
CLINICAL CASE 1 (Continued)
The patient has a negative PET/CT scan and elects close observation. She is tired of frequent BM biopsies and complex imaging and is inquiring about more sensitive blood-based assays to follow her disease.
CLINICAL CASE 2 (Continued)
The patient is happy to hear he is now MRD negative. However, he still has detectable IgG kappa in his serum and this concerns him. He read that daratumumab is detectable as an IgG kappa by conventional methods and wants to know more about the ability of Matrix-assisted laser desorption/ionization- time-of-flight (MALDI-TOF) mass spectrometry in resolving daratumumab interference.
What is the role of novel blood-based assays in characterizing circulating monoclonal proteins?
NGF and NGS approaches used for BM assessment are not accurate enough for MRD detection in the peripheral blood. Mass spectrometry (MS)–based approaches to evaluate for circulating monoclonal proteins are now used by many institutions and have several benefits over traditional IFE. MALDI-TOF assays are the most popular given their high throughput and low cost28,29 ; however, their sensitivity is not yet high enough for them to be used for peripheral blood MRD assessment. This assay can identify intact monoclonal proteins (Figure 1) with greater sensitivity compared to IFE. It is not yet as sensitive as serum-free light chain testing when considering light chain-only cases. As a result, it currently should be considered a method that could be ordered instead of IFE that has several benefits, as we highlight below. In 1 study an additional 6% of IFE-negative cases overall were reclassified as positive after MALDI-TOF,30 and as many as 12% were when considering amyloid light chain (AL) amyloidosis.31 In the case of low burden states, such as precursor disease (monoclonal gammopathy of indeterminate potential), MALDI-TOF can identify approximately twice as many positive cases compared to IFE.32,33 It remains to be determined what the natural history and clinical significance of precursor states identified only using novel, sensitive assays, termed monoclonal gammopathy of indeterminate potential,33 will be.
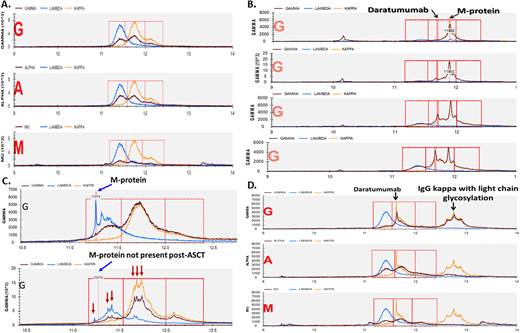
(A) MALDI-TOF results in normal serum (no monoclonal protein present). The x-axis represents mass/charge ratio. Three separate rows are presented for each heavy chain isotype. (B) MALDI-TOF results after each cycle of daratumumab in a patient with IgG kappa MM. The pathogenic clone and daratumumab have different molecular weights and are therefore easily distinguishable. Reference mass/charge ratios exist for several other moAbs used in treating myeloma that can achieve a serum concentration high enough to cause interference. (C) “Immune reconstitution” (oligoclonal) pattern seen after autologous transplant. Multiple IgG kappa and lambda clones are noted, but the initial pathogenic clone is absent. (D) A monoclonal IgG kappa protein with a glycosylated light chain in an AL amyloidosis patient treated with daratumumab. Glycosylated IgG kappas are up to 12 times more common in patients with AL amyloidosis and are present years before the diagnosis of AL amyloidosis is made.
Used with permission from Dr David Murray.
(A) MALDI-TOF results in normal serum (no monoclonal protein present). The x-axis represents mass/charge ratio. Three separate rows are presented for each heavy chain isotype. (B) MALDI-TOF results after each cycle of daratumumab in a patient with IgG kappa MM. The pathogenic clone and daratumumab have different molecular weights and are therefore easily distinguishable. Reference mass/charge ratios exist for several other moAbs used in treating myeloma that can achieve a serum concentration high enough to cause interference. (C) “Immune reconstitution” (oligoclonal) pattern seen after autologous transplant. Multiple IgG kappa and lambda clones are noted, but the initial pathogenic clone is absent. (D) A monoclonal IgG kappa protein with a glycosylated light chain in an AL amyloidosis patient treated with daratumumab. Glycosylated IgG kappas are up to 12 times more common in patients with AL amyloidosis and are present years before the diagnosis of AL amyloidosis is made.
Used with permission from Dr David Murray.
MALDI-TOF provides the following additional benefits over IFE: (1) it can resolve monoclonal antibody (moAb) interference, (2) it can identify glycosylated light chains, which are more likely to be associated with AL amyloidosis and cold agglutinin disease, and (3) it can differentiate between pathogenic clones and nonpathogenic clones. Currently, the daratumumab IFE reflex assay is commonly used to establish whether a residual IgG kappa serum protein in patients treated with daratumumab is due to residual disease or the drug itself. However, several moAbs used to treat MM (daratumumab, elotuzumab, isatuximab, belantamab, and several bispecific antibodies) reach high enough concentrations in the serum to become detectable by IFE. MALDI-TOF can differentiate between a moAb and a pathogenic clone in 84% of cases (Figure 1).34 The remaining cases may need to be reflexed to higher-resolution MS assays since the mass of the moAb may be too close to that of the pathogenic clone. Similarly, MALDI-TOF can differentiate between benign, oligoclonal, immune reconstitution patterns, and reappearances of the pathogenic clone.35 This phenomenon can occur in up to 22% of patients, especially after ASCT, and is associated with improved outcomes.36 Finally, glycosylated light chains (Figure 1), especially kappa, are associated at a much higher rate with rare dysproteinemias such as AL amyloidosis and cold agglutinin disease and can be present years before diagnosis.37-39
BM MRD and MALDI-TOF were independently prognostic of PFS and OS in the STAMINA trial and in single-center studies post ASCT.40,41 However, more studies are required to evaluate the incremental prognostic value of these assays over BM MRD and traditional IFE. Finally, MS methods pairing high-sensitivity mass spectrometers with liquid chromatography are more sensitive than MALDI-TOF and may have a role in MRD detection in the peripheral blood.42
Circulating tumor cell (CTC) detection in the peripheral blood using NGF also appears to be a powerful prognostic marker in assessing disease outcomes. CTCs were evaluated in the PETHEMA/GEM2012MENOS65 trial. Each log increase of CTCs was roughly associated with at least a 10% absolute decrease in 5-year PFS rates,43 although this association was not as consistent with OS. CTCs were predictive of PFS in patients with MRD-positive CR but not those in MRD-negative CR. Similar results were demonstrated in CTC analysis from patients treated in the FORTE trial,44 where CTCs were predictive of both PFS and OS regardless of traditional prognostic factors and hematologic response but not MRD-negative CR. Additional studies at various time points (eg, relapse disease, frail patients) will help clarify the role of CTC evaluation in MM prognosis. Other approaches, like assessing circulating tumor DNA, are promising but require additional validation.
Therefore, we can inform patient 1 that MS-based approaches used in clinical practice are more sensitive than IFE and could be useful to detect an early biochemical relapse. We can inform patient 2 that MALDI-TOF can easily resolve daratumumab interference, assuming that his IgG kappa has a different mass to that of daratumumab.
Conclusions
Novel laboratory and imaging methods to evaluate disease burden provide an unprecedented ability to discriminate and substratify MM patients who achieve deep conventional responses. Unfortunately, there is as yet no evidence to support individualizing therapy based on the results of these studies. This is currently done in clinical practice but only after carefully discussing it with patients and considering MM risk and patient frailty. In our institution and outside of clinical trials, MRD testing is routinely performed in all patients without morphologic or immunophenotypic (“low-resolution” flow cytometry) evidence of BM involvement by myeloma after ASCT or chimeric antigen receptor T-cell therapies and to discuss prognosis with the patient. There is no consensus on the optimal subsequent time points for BM MRD assessment or what to do with the result. Therefore, these are not performed routinely but rather on a case-by-case basis and after discussion with the patient about the impact of these findings on prognosis and management. However, this may soon change since several studies (Table 2) are currently evaluating strategies to adjust therapy according to findings from these more sensitive methods.
Phase 3 trials using MRD status to guide intensification or deintensification of therapy
| Title | Intervention/description | Phase | Estimated primary completion date | Primary end point | Brief outline |
|---|---|---|---|---|---|
| NCT04071457 DRAMMATIC study | Drug: lenalidomide Drug: daratumumab/rHuPH20 | Phase 3 | 1 July 2029 | Overall survival | After 2 years of maintenance, MRD+ patients continue with the assigned treatment. MRD− patients are randomized to continue/discontinue treatment. |
| NCT04513639 REMNANT study | Drug: early treatment of relapse with carfilzomib, dexamethasone, daratumumab Drug: standard treatment of relapse with carfilzomib, dexamethasone, daratumumab | Phase 2-3 | 1 June 2030 | PFS, OS, MRD negativity after first-line treatment | Newly diagnosed patients are treated with standard induction. Patients that reach MRD negativity are randomized to early treatment after conversion to MRD+ vs treatment after progression as defined by IMWG. |
| Title | Intervention/description | Phase | Estimated primary completion date | Primary end point | Brief outline |
|---|---|---|---|---|---|
| NCT04071457 DRAMMATIC study | Drug: lenalidomide Drug: daratumumab/rHuPH20 | Phase 3 | 1 July 2029 | Overall survival | After 2 years of maintenance, MRD+ patients continue with the assigned treatment. MRD− patients are randomized to continue/discontinue treatment. |
| NCT04513639 REMNANT study | Drug: early treatment of relapse with carfilzomib, dexamethasone, daratumumab Drug: standard treatment of relapse with carfilzomib, dexamethasone, daratumumab | Phase 2-3 | 1 June 2030 | PFS, OS, MRD negativity after first-line treatment | Newly diagnosed patients are treated with standard induction. Patients that reach MRD negativity are randomized to early treatment after conversion to MRD+ vs treatment after progression as defined by IMWG. |
IMWG, International Myeloma Working Group.
MRD negativity is increasingly being used as an end point in several clinical trials; however, some challenges remain (Table 3). Current and future trials will help answer key questions: (1) Should therapy be escalated or deescalated based on MRD results? (2) Can MRD negativity be used as a surrogate early end point for OS in clinical trials? (3) What time points are optimal for MRD testing according to disease setting (newly diagnosed vs relapsed disease)? (4) What is the best method, or even combination of methods, to use? Finally, a key question lingers: Why do patients with no evidence of disease, despite the use of novel detection methods, relapse? Is it really a pool of plasma cells that remain undetectable at such low levels that gives rise to relapsed disease? Is the source of relapse phenotypically or genomically very different from what we typically consider a malignant plasma cell, and is a BM aspiration the best way to sample these cells? In other words, are we looking for the right cell and using the right method? The study of these patients and the answers to these questions bring us closer to achieving a cure for MM.
Barriers and questions for the regular use and future prospects of MRD use in MM
| 1. What is the most appropriate sensitivity threshold (10−5,10−6, or higher) to determine MRD presence? |
| 2. Should MRD− cutoffs or requirement for sustained MRD− be different according to disease risk? |
| 3. What is the optimal timing for MRD assessment? What are the optimal intervals for sustained MRD assessment? |
| 4. Can clinicians intensify or deintensify their therapeutic approaches based on MRD results at different time points? |
| 5. How can blood-based and imaging methods complement BM-based MRD assessment? |
| 6. Can MRD− be used as a surrogate marker for more clinically relevant end points (ie, PFS and ideally OS)? If yes, how “much more” MRD− is needed for a therapy to consistently lead to improve PFS/OS and in which setting (newly diagnosed vs relapsed disease, high-risk disease vs not)? |
| 7. Are there tumor-extrinsic factors that can explain early relapse in MRD– non–high-risk patients (immunome, microbiome)? |
| 8. What cells are responsible for relapse in MRD−? Are they malignant plasma cells truly present at very low thresholds, or are they phenotypically and genomically different than plasma cells? Are they amenable to sampling by BM aspiration, or are they adherent to the BM niche? |
| 1. What is the most appropriate sensitivity threshold (10−5,10−6, or higher) to determine MRD presence? |
| 2. Should MRD− cutoffs or requirement for sustained MRD− be different according to disease risk? |
| 3. What is the optimal timing for MRD assessment? What are the optimal intervals for sustained MRD assessment? |
| 4. Can clinicians intensify or deintensify their therapeutic approaches based on MRD results at different time points? |
| 5. How can blood-based and imaging methods complement BM-based MRD assessment? |
| 6. Can MRD− be used as a surrogate marker for more clinically relevant end points (ie, PFS and ideally OS)? If yes, how “much more” MRD− is needed for a therapy to consistently lead to improve PFS/OS and in which setting (newly diagnosed vs relapsed disease, high-risk disease vs not)? |
| 7. Are there tumor-extrinsic factors that can explain early relapse in MRD– non–high-risk patients (immunome, microbiome)? |
| 8. What cells are responsible for relapse in MRD−? Are they malignant plasma cells truly present at very low thresholds, or are they phenotypically and genomically different than plasma cells? Are they amenable to sampling by BM aspiration, or are they adherent to the BM niche? |
Conflict-of-interest disclosure
Matthew Ho: no competing financial interests to declare.
Taxiarchis Kourelis: no competing financial interests to declare.
Off-label drug use
Matthew Ho: nothing to disclose.
Taxiarchis Kourelis: nothing to disclose.
