
Abstract
The thalassemias are inherited quantitative disorders of hemoglobin synthesis with a significant worldwide burden, which result in a wide spectrum of disease from the most severe transfusion-dependent form to the mildest asymptomatic carrier state. In this article, we discuss the importance of carrier, prenatal, and newborn screening for thalassemia. We examine the rationale for who should be screened and when, as well as the current methodology for screening. Deficiencies in the newborn screening program are highlighted as well. With the advent of inexpensive and rapid genetic testing, this may be the most practical method of screening in the future, and we review the implications of population-based implementation of this strategy. Finally, a case-based overview of the approach for individuals with the trait as well as prospective parents who have a potential fetal risk of the disease is outlined.
Learning Objectives
Understand the current guidelines and recommendations for screening and diagnosis of thalassemia syndromes
CLINICAL CASE
A 24-year-old woman of Thai descent is planning to start a family with her partner, who is of Chinese descent. She recalls being told she is anemic all her life and has been on iron supplements off and on for many years, without correction of her anemia. She recalls that her mother has the same medical history. Her internist refers her to hematology for testing, and she is diagnosed with β-thalassemia trait. She is then referred to genetics with her partner (a recent immigrant who has never been tested) for testing and counseling regarding the risk of thalassemia in their offspring.
Introduction
The thalassemias are a group of genetic diseases with a high prevalence and significant morbidity. The broad range of clinical manifestations and complications, as well as high burden of disease, from the quality-of-life as well as financial standpoint, underscores the importance of minimizing its prevalence and optimizing outcomes in those who are affected, through early diagnosis and comprehensive care. Identification of individuals with mild disease or carriers, prenatal screening, and counseling are key to increasing awareness of the disease, with the potential for reducing transmission. Current newborn screening identifies affected individuals with the more common, but not all, severe α- and β-thalassemias, but most carriers may not be identified in this manner. In time, with advances in technology for genetic testing, the platform for screening may include direct gene analysis using diagnostic chips or whole-genome or exome sequencing, providing a more complete genetic picture and allowing for more definitive carrier identification. A brief overview of pathophysiology and a rational approach to testing and screening are examined based on the why, who, when, and how.
Pathophysiology
Several hundred mutations have been described in the α- and β-globin gene clusters, and the resulting imbalance between the complementary globins results in intramedullary apoptosis of the maturing erythroid precursors and leads to the hallmark ineffective erythropoiesis (IE).1 These genotypes are summarized in Figure 1.

Mutations or deletions of the α-globin genes result in an excess of γ-globin in the fetus. When all 4 α-genes are deleted, HbF cannot be produced in the second trimester and results in severe anemia and hydrops fetalis. When 3 genes are deleted or mutated, there is formation of tetramers of γ-globin (hemoglobin Barts [HbBarts]) initially and β-globin (HbH) later. When both β-globin genes are mutated, there is decreased or absent HbA production in the third trimester and progressive anemia postnatally. The characterization of compound heterozygotes with β-thalassemia and HbE mutations leads to a similar picture but with the additional presence of HbE.
As a result of IE and anemia, clinical manifestations vary, with the most severely affected individuals requiring lifelong regular transfusions and iron chelation, with potential for a variety of complications.1,2 Less severely affected individuals may not require regular transfusions but may still develop manifestations secondary to the IE such as extramedullary hematopoiesis, bone changes, and vascular disease, also leading to significant morbidity.1,2 Treatment and monitoring are cumbersome, are expensive, and significantly affect quality of life. Thus, the thalassemia syndromes have a heavy burden of disease and are good candidates for screening.
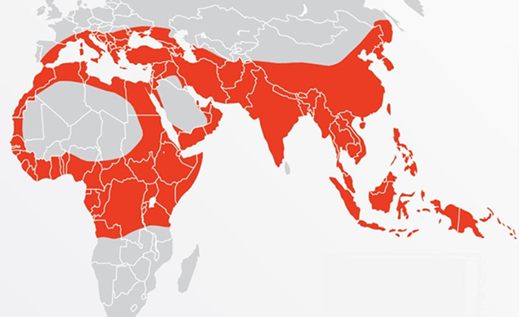
Scope of the problem
About 1.5% of the world's population carries a thalassemia gene mutation, approximating 80 to 90 million people worldwide (Figure 2).3,4 The carrier state is found in Africa, around the Mediterranean, and the Middle East, but most affected individuals live in the Indian subcontinent, Southeast Asia, and China.1,3,4 However, changing immigration patterns have led to a worldwide distribution of the disease.5
Why we should screen
Screening for thalassemia syndromes is important in 3 main settings: for the correct diagnosis and management of anemia, prenatal counseling for parents at risk of having an affected child, and newborn screening for early diagnosis.6
Thalassemia carriers are commonly misdiagnosed with iron-deficiency anemia and often receive prolonged courses of iron, occasionally parenterally. Appropriate screening would obviate unnecessary use of iron in carriers and monitoring prevents sequelae and chronic complications in those with thalassemia intermedia.
Prenatal screening can be used to identify adult carriers interested in starting families. For known carriers, screening of partners leads to prenatal risk assessment and family planning counseling. Pregnant women diagnosed with thalassemia trait should have partner screening to assess fetal risk, particularly for fetuses with hydrops fetalis and stillbirth, and receive education and counseling.7 Earlier diagnosis of hydrops fetalis and stillbirth can mitigate the mental and physical consequences for pregnant mothers.
Newborn screening programs allow early identification of affected infants, who can be monitored closely for the development of anemia, and start transfusions at the appropriate time, minimizing the risk for complications.
Who should be screened
Individuals—based on demographics and history
Adults and children of Asian, African, or Mediterranean ancestry, with a family history of thalassemia, family history of lifelong anemia, and those with a history of microcytic anemia unresponsive to iron supplementation or microcytic anemia without iron deficiency, should be screened for thalassemia.6
Prospective parents—prenatal screening
The American College of Obstetrics and Gynecology (ACOG) recognizes that patients with African, Mediterranean, and Southeast Asian ancestry are at increased risk for hemoglobinopathies, including sickle cell and thalassemia.7 As a result of more widespread mixing of populations, limiting screening to these ancestries may not be sufficient, and broader guidelines are necessary.8
Prospective parents who are identified as thalassemia carriers should receive prenatal genetic testing with counseling. If one partner is confirmed to be a carrier, the other should be extensively tested. Couples who are both thalassemia carriers can be offered in vitro fertilization and preimplantation genetic diagnosis.7 For expectant parents, antepartum genetic testing can be offered prior to delivery along with counseling.
Newborns
At-risk newborns should be screened for thalassemia syndromes.6 However, this poses technical challenges because the methodology currently used for newborn hemoglobinopathy screening (in all 50 states in the United States) is specifically designed to detect sickle hemoglobin. The detection of HbBarts (tetramers of γ-globin) on the newborn screen would facilitate the early diagnosis of α-thalassemia trait or HbH disease, but this requires hemoglobin quantitation, not performed universally. The most severe form of β-thalassemia, β0/β0-thalassemia, could be diagnosed by the absence of HbA on the newborn screen. Less severe forms of β-thalassemia could also be detected but not usually the trait. In regions where thalassemia is more common, particularly Asia, implementing newborn screening methodology to be able to detect thalassemia would be an important strategy for early diagnosis, counseling, and regular comprehensive care where appropriate.
Infants
The American Academy of Pediatrics had a longstanding recommendation for a screening hemoglobin/hematocrit for all infants at 9 months of age.9 To prevent iron deficiency, iron-fortified formula is now recommended, and screening is done at 12 months of age for formula-fed infants. Severe thalassemia, β0/β0-thalassemia, usually presents well before this age with progressive anemia and other clinical manifestations. However, more intermediate forms, including β+/β+-thalassemia, HbE/β0-thalassemia, or HbH disease, may have few clinical manifestations besides moderate anemia and may be missed until the 9- to 12-month hemoglobin/hematocrit is done. Screening these infants would be important to establish a diagnosis, preclude unnecessary iron supplementation, and begin monitoring and comprehensive care to optimize outcomes and minimize morbidity.
How we screen
Newborn thalassemia screening
With the high prevalence and wide distribution of hemoglobinopathies, testing for these was incorporated into the newborn screen.6 The normal newborn at term has approximately 80% to 90% HbF and 10% to 20% HbA, with trace amounts of other hemoglobins such as HbA2, which are typically not reported.
α-Thalassemia newborn screening
α-Thalassemia syndromes compatible with life can be detected on newborn screening. Fetuses with 3 α-gene deletions or 2 α-gene deletions and 1 α-gene with a Constant Spring mutation (or other nondeletional α mutation) will produce excess unbound γ-globin, which will result in the formation of γ-tetramers, designated HbBarts, identifiable on newborn screening. The amount of HbBarts correlates with the severity of α-thalassemia, although not in all instances, and can be detected antepartum and at birth.9,10 Newborns with 2 α-gene deletions may have 3% to 6% HbBarts at birth, and a silent carrier missing only 1 α-gene may have 1% to 4% at birth.11
β-Thalassemia newborn screening
Fetuses with β0/β0 mutations will produce no HbA and will have only HbF on the newborn screen, only otherwise seen in extreme prematurity, where β-globin expression has not begun yet. Thus, gestational age is an important consideration, and normal ranges should be established at various gestational ages for easier interpretation. Fetuses with 1 β0-mutation and 1 β+-mutation or 2 β+-mutations will have some HbA on the newborn screen. If this is quantified, it is less than the normal range—approximately 3% to 5%—and may be flagged as possible β-thalassemia.6 Individuals with β0/HbE may have no HbA and only have HbF and HbE, whereas those with β+/HbE will have some HbA. HbA2, which is elevated in carriers, is produced in very small quantities in the fetus (0.5%) and is not typically reported on the newborn screen.1,2
Newborn screening in the United States
There is currently no standard for newborn screening across the United States, and recommendations for testing vary from state to state.12 In 2017, the American Academy of Pediatrics reviewed newborn hemoglobinopathy screening across the 50 states.13 Thirty-one states recommended anemia screening beyond routine care or sickle cell screening. Five of the 31 states recommended hemoglobin electrophoresis and 4 recommended hematology referral.13 Only 1 recommend initial genetic testing. The other 19 states do not have thalassemia screening recommendations beyond routine care. Twelve of these 19 states reported α-thalassemia trait with no additional follow-up, and 7 did not report α-thalassemia or carrier status at all.13
Initial newborn screening in most states is by high-performance liquid chromatography (HPLC) or isoelectric focusing (IEF) on a spot of blood. These tests detect hemoglobin variants and may provide limited data to support diagnosis of a thalassemia syndrome. HPLC, either by cation exchange or reverse phase, quantifies hemoglobin variants such as hemoglobin A, A2, F, S, H, and Barts.13,14 However, different hemoglobin variants may coelute, making it difficult to distinguish one variant from another. Thus, HPLC is used for initial screening and is followed by a confirmatory test, either IEF or molecular testing.10 IEF is more labor intensive but identifies and quantifies hemoglobin variants more precisely than standard hemoglobin electrophoresis.13 Molecular testing includes polymerase chain reaction (PCR)–based testing or DNA sequencing.1 Allele-specific PCR-based methods are used to detect point mutations in α- and β-globin genes. Gap PCR is used to detect deletions, α duplications, and deletional hemoglobin variants.1 DNA sequencing is more expensive and more comprehensive and can be used to diagnose previously unidentified mutations.1
There is no nationwide standard of reporting HbBarts percentage to providers, patients, or their families.10 A Centers for Disease Control and Prevention survey found that HbBarts reporting varies among newborn screening programs, and some programs do not report quantitative HbBarts or recommend follow-up.13
Thalassemia screening in children and adults
Prospectively screening all patients for thalassemia is neither cost-effective nor indicated because iron deficiency is relatively much more common and explains a microcytic anemia. Establishing the diagnosis of thalassemia trait is relevant, and avoidance of unnecessary iron supplementation is important. Individuals with an intermediate-severity thalassemia syndrome with IE have complications that could be prevented if comprehensive care is provided early.
In the following situations, screening for thalassemia is recommended:
Family history of thalassemia—trait or disease
Microcytic anemia with a negative history for iron deficiency— adequate dietary iron intake, absence of blood loss
Persistent microcytic anemia despite an adequate trial of supplemental iron
Testing of such individuals can be approached in a stepwise manner (Figure 3).
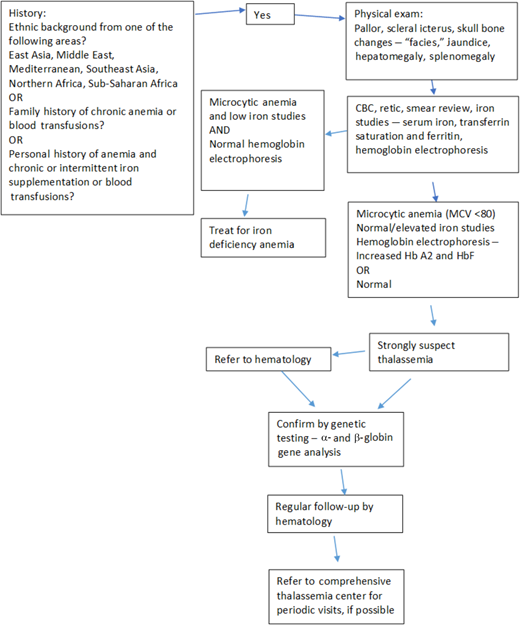
Ideal algorithm for thalassemia screening, developed to screen for thalassemia syndromes in adults based on history, physical examination, and laboratory parameters.
Ideal algorithm for thalassemia screening, developed to screen for thalassemia syndromes in adults based on history, physical examination, and laboratory parameters.
Prenatal screening
ACOG recommends that women with a low mean corpuscular volume (MCV) should have serum ferritin assessment. Those with normal levels and microcytic anemia should have hemoglobin electrophoresis testing.7 For patients with a normal hemoglobin electrophoresis and Asian ancestry, genetic testing for α-thalassemia is recommended. In each situation, partner testing is key to determining fetal risk and is based on hemoglobin electrophoresis or genetic testing as appropriate for β- and α-thalassemia, respectively.8 Couples who are both identified as carriers should receive genetic counseling to determine risk of an affected fetus, and family decision support should be provided. The most severe form of α-thalassemia—deletion of all 4 α-genes—results in hydrops fetalis, with severe anemia (hemoglobin ranging from 3-8 g/dL), organomegaly, and edema, usually leading to intrauterine fetal demise from heart failure, with preterm labor, stillbirth, and a detrimental effect on maternal health.1,11 Prenatal screening would identify the at-risk fetus, and amniocentesis or chorionic villus sampling would confirm the diagnosis.11 In a suspected pregnancy, Doppler assessments of cerebral vessel flow velocities or direct fetal blood sampling to quantify the anemia could be used. If intrauterine transfusions are instituted in time, such fetuses may survive but would be transfusion dependent postnatally— designated α-thalassemia major.11
It is important to keep in mind that 2 α-gene deletions are not uncommon in African ancestry individuals, but these mutations tend to occur on opposite chromosomes (trans configuration), and hence the risk of having a fetus with all 4 genes deleted is lower than in individuals with East Asian ancestry, in whom the 2 gene deletions are more commonly on the same chromosome (cis configuration). For women who are identified as having β-thalassemia trait or HbS trait, partner testing of hemoglobin fractionation is imperative to assess fetal risk for β-thalassemia or sickle cell disease (HbS-β thal).1
Preimplantation genetic testing
Preconception, ACOG recommends genetic testing and counseling for couples at high risk for thalassemia as noted.7 For couples interested in avoiding elective termination, in vitro fertilization and preimplantation genetic diagnosis can be offered.7 For couples who were not diagnosed as high risk for thalassemia prior to pregnancy, DNA testing via chronic villus sampling or amniocentesis should be offered. Counseling should be offered in all such situations.7 This approach has resulted in a major reduction in the birth rate of infants with thalassemia throughout the Mediterranean region and the Middle East, as well as in parts of the Indian subcontinent and Southeast Asia. Novel approaches continue to be explored in an attempt to avoid the use of invasive procedures such as chronic villus sampling and amniocentesis, including harvesting fetal DNA from fetal cells in maternal blood or maternal plasma.15,16
Suggested approach for screening of individuals at risk, all ages
As the demographics continue to change globally and in the United States, a comprehensive approach for early diagnosis will improve the health of patients and minimize risk. We suggest the following, starting preconception and through adulthood:
Screening and identification of thalassemia carrier status and counseling early for all prospective mothers at risk based on ethnicity, family history, medical history, and laboratory parameters. Confirmatory genetic testing of both partners and genetic counseling must be offered and, if available, preimplantation genetic diagnosis when appropriate.
If a pregnancy has already occurred in a mother at risk, the same screening should be done, and if thalassemia trait is confirmed as described previously, partner testing and genetic counseling are indicated. Testing of the fetus should be offered if both parents are carriers or if the father's status cannot be ascertained. More advanced techniques such as fetal DNA in maternal blood need to be tested more extensively to avoid more invasive testing procedures. If the fetus is found to be affected, appropriate counseling should be provided.
Newborn screening should be standardized to include hemoglobin characterization and fractionation for all. Ideally, as genetic testing becomes more widely available, technically easier to perform, and cheaper, moving testing to a genome/exome or chip technology would be most effective in definitively identifying individuals with thalassemia. We recognize this may be a future-oriented goal and a not widely feasible approach. Until universal genetic testing can be performed, we recommend the optimal strategy of performing a hemoglobin electrophoresis, HPLC, or IEF on all infants; reporting the hemoglobin variants, especially HbBarts, and unstable hemoglobin variants; and quantifying HbA. Additions to the newborn screen are justified if a treatment introduced early would reduce morbidity and mortality. For thalassemia, this remains controversial, and prospective data demonstrating the benefits of this approach could provide a rationale.
We recommend screening all individuals (adults and children) with appropriate histories, certain physical examination findings, and microcytic anemia for thalassemia using the algorithm in Figure 3.
CLINICAL CASE (continued)
The couple is at risk for an affected child given that the prospective mother has β-thalassemia trait, and the prospective father is of Chinese descent. He should have carrier testing for β-thalassemia trait. If he is a carrier, then the potential fetus is at risk of having the disease. Preimplantation diagnosis should be offered before conception, or testing of the fetus should be offered early in the pregnancy. Some families may choose neither option.
Conclusion
Due to increasingly changing demographics and migration from areas where thalassemia is more prevalent to those where it is less so, the prevalence of thalassemia is changing. It has become an important health issue to identify patients with thalassemia syndromes and thalassemia trait to be able to not only provide early comprehensive care but also avoid unnecessary interventions. Early diagnosis can prepare at-risk couples, as well as identify at-risk fetuses and newborns. Newborn screening for hemoglobin disorders, pioneered with a sickle cell focus, has come a long way, but there is still room for advances to achieve the goal of optimal screening for thalassemia and establishment of an early diagnosis to optimize management.
Acknowledgments
C.M and S.S. have received funding from the Centers for Disease Control and Prevention and the Health Resources and Services Administration.
Conflict-of-interest disclosure
Cheryl Mensah has served as a consultant to Bluebird bio and Chiesi. She currently serves on adjudication committee for CRISPR/Vertex for thalassemia.
Sujit Sheth is a consultant to Agios, Bluebird bio, Bristol Myers Squibb, and Chiesi and serves on a clinical trial steering committee for CRISPR/Vertex CTX001 for thalassemia.
Off-label drug use
Cheryl Mensah: nothing to disclose.
Sujit Sheth: nothing to disclose.
