

Abstract
Oral hypomethylating agents (HMAs) represent a substantial potential boon for patients with myelodysplastic syndrome (MDS) who have previously required between 5 and 7 visits per month to an infusion clinic to receive therapy. For patients who respond to treatment, ongoing monthly maintenance visits represent a considerable burden to quality of life, and for those who are early in therapy, these sequential visits may tax transportation and financial resources that would be optimally distributed over the treatment cycle to facilitate transfusion support. The availability of oral HMAs may support the optimal application of these agents by contributing to adherence and lessening the burden of therapy, potentially encouraging patients to stay on longer-term treatment. Distinct pharmacokinetic profiles for the recently approved oral HMAs (oral azacitidine and decitabine-cedazuridine) result in differential toxicity profiles and have prompted their clinical trial development in lower- and higher-risk MDS, respectively.
Learning Objectives
Describe the challenges of effective therapy with HMAs for patients with MDS and the barriers to this therapy
Understand the PK profiles for HMAs given parenterally and contrast these with the PK profiles following oral administration of unmodified oral azacitidine and decitabine/cedazuridine
Recognize how the PK for oral HMAs may differ from an established parenteral regimen and anticipate the challenges associated with transitioning from one regimen to another
CLINICAL CASE 1
A 78-year-old woman presented to our clinic after routine blood work demonstrated thrombocytopenia and neutropenia. The patient was otherwise in reasonable health for her age with comorbid medically managed hypertension. She was widowed but lived independently, with minimal support from her local adult children. A complete blood count (CBC) at the initial evaluation demonstrated a white blood cell (WBC) count of 1.4 × 109/µL, with an absolute neutrophil count of 0.5 × 109/µL, hemoglobin (Hg) of 11 g/µL, and platelet count of 25 k/µL. She was demonstrated to be nutrient replete for B12, folate, iron, and copper. A bone marrow (BM) aspiration and biopsy were performed, showing refractory anemia with excess blast-2 (15% blasts); cytogenetics were normal. Next-generation sequencing (NGS) revealed mutations in IDH2 and SRSF2. A diagnosis of higher-risk myelodysplastic syndrome (MDS) was rendered (International Prognostic Scoring System [IPSS]-Revised [R], 5.5 points [high risk]; IPSS 2.0 points [intermediate-2]), and the patient started therapy with parenteral azacitidine 75 mg/m2 given subcutaneously (SQ) 7 days of a 28-day schedule. The initial treatment was characterized by the development of red blood cell (RBC) and platelet transfusion dependence in cycles 1 and 2. She presented prior to cycle 3 of therapy, and the CBC showed a WBC count of 1.0 × 109/µL with absolute neutrophil count 0.2 × 109/µL, an Hg of 8 g/µL, and platelets, 105 k/µL. She came to the clinic accompanied by her children. She stated that she was no longer willing to continue therapy. She reported intolerable loss of quality of life and independence due to the daily infusion clinic visits in the first week of each cycle and the burden of transfusion dependence. She was adamant that she would take no further chemotherapy despite an early response to treatment that suggested she was likely to derive a survival benefit from continued treatment (early hematologic improvement [HI] of the platelet lineage). She requested enrollment in home hospice care and passed away 1 month after therapy discontinuation. Today, the availability of oral agents might have convinced this patient to continue treatment, optimizing her survival and improving her quality of life. As it stood, this patient accrued all the toxicity from the use of this agent, including initial loss of quality of life, without realizing the longer-term potential benefits of transfusion independence (TI) and survival, which she might have been expected to achieve given her early evidence of response.
Introduction
Hypomethylating agents (HMAs) have been the mainstay of treatment for patients with higher-risk MDS since the initial regulatory approval of monthly treatments with parenteral azacitidine and decitabine in the early 2000s.1 The shared active metabolite of both agents (~15% of the administered azacitidine dose, converted by the action of ribonucleotide reductase) is decitabine triphosphate, an analogue of the nucleoside cytidine (Figure 1). Although 85% of parenterally administered azacitidine is incorporated into RNA, interrupting protein synthesis and inducing apoptosis, the clinical impact of this incorporation on its activity remains an area of active investigation.1 When decitabine triphosphate is incorporated into DNA whose parent strand bears a methyl group in the context of a cytosine-phospho-guanine sequence, DNA methyltransferase enzymes (particularly DNMT-1) bind it irreversibly, creating an adduct that results in both DNA damage and the depletion of DNMT enzymes.2 With repeated cycles of DNA replication, loss of the methylation enzymes results in the passive demethylation of DNA at multiple loci. At low doses that do not cause overwhelming cytotoxicity, these drugs induce transient gene-specific and global hypomethylation that are hypothesized to mediate their response. Historically, activity has been postulated to result from the reexpression of epigenetically silenced tumor suppressor genes, but more recently, reexpression of endogenous retroviral elements with enhanced immune recognition of MDS progenitors has been hypothesized as another putative mechanism.3 Despite ongoing controversy over the definitive explanation for therapeutic efficacy, optimal patient outcomes from HMA therapy seem to require long-term treatment on a regular and uninterrupted schedule.4-7
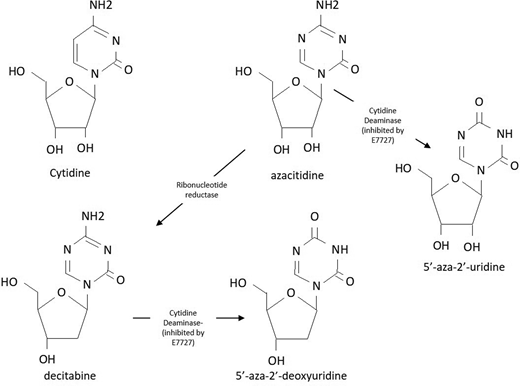
Chemical structure of cytidine, azacitidine analogues, and the breakdown products of CDA.
Chemical structure of cytidine, azacitidine analogues, and the breakdown products of CDA.
Dose, schedule, and treatment continuation are critical to optimal HMA response
When given on a monthly basis for repeated cycles, these agents improve cytopenias, lower BM blasts, improve quality of life, decrease transformation to acute myeloid leukemia (AML), and improve survival for select patients.8-10 Despite years of study, no effective pretreatment predictors of response are clinically applicable.11 About half of HMA-treated patients will respond, most with improvements in cytopenias (HI of the affected lineage) and a minority (perhaps 15%) with complete response (CR), but responses can take several months to develop (National Comprehensive Cancer Center guidelines recommend a minimum of 6 cycles of treatment before response assessment) and are generally of limited durability.12,13 Critically, hematological improvement (HI) in any lineage is associated with survival benefit for patients who continue therapy; with sustained treatment some individuals will have improved blood counts across more than one lineage, even converting to CR.7,13,14 Unfortunately, when patients discontinue therapy, rapid progression and relapse with poor survival are likely.15-21 Good responses and improved survival are often accompanied by recovery of the platelet count in the first 1 to 3 cycles of treatment, an early biomarker for those who are likely benefiting from treatment.22,23
As for the patient in our first case, early cycles of HMA therapy are often characterized by the worsening of cytopenias and the development of transfusion dependence. This has the potential to create a substantial burden for patients and families, especially since each monthly cycle has historically required 5 to 7 consecutive daily doses of intravenous (IV; decitabine, azacitidine) or SQ (azacitidine) treatment at a designated infusion center. Given the early worsening of cytopenias, optimal supportive care requires weekly or even twice-weekly visits during the subsequent 3 weeks to assess transfusion needs as well as the prophylactic use of antimicrobials.24 The development of RBC and platelet transfusion dependence and neutropenia early in the course of treatment can trigger clinicians to inappropriately reduce doses, delay, or even discontinue therapy; decisions known to compromise efficacy.25,26 Such decisions may explain the difference between the survival benefit documented in controlled populations of patients treated with HMAs in the context of clinical trials compared to outcomes in unselected populations of patients, where improved survival rates have been less consistent.10,21,27,28
A set of recent retrospective analyses using the surveillance, epidemiology, and end results Medicare-linked database in a large cohort of 644 patients with higher-risk MDS evaluated between 2011 and 2015 sought to investigate the way in which patients treated in a real-world setting routinely receive therapy. This study demonstrated that almost 30% of patients discontinued HMAs before completing 4 cycles—a majority after completing only 1 cycle of treatment.29 A more detailed analysis of this same cohort reveals that an additional 20% of patients had early dose delays of >90 days between cycles, which likely obviates response.30 Overall, among the approximately 50% of patients with higher-risk MDS, those who did not receive their HMA on schedule for at least 4 cycles had substantially higher rates of health care utilization characterized by increased emergency room visits, higher rates of hospitalization, and more admissions to skilled nursing facilities and hospice. Suboptimally treated patients in this cohort were more likely to be older and unpartnered.
Real-world studies further demonstrate the relatively limited initiation of HMA treatment for high-risk patients, with only 12% to 30% of presumably eligible patients receiving therapy at all, depending upon the report.20,27,30 These data highlight the fact that despite the availability of HMAs as primary therapy for MDS for more than 15 years, many patients do not receive any therapy at all, and among those who do, early discontinuation or inadequate dose intensity during early cycles probably compromises response.30 Such treatment decisions are likely to have an adverse impact on quality of life and compromise survival for those MDS patients, who will accrue all the toxicity from HMA therapy and none of the benefits (HI, CR, or survival) of sustained therapy.24 The importance of adequate support and therapy persistence for patients with higher-risk MDS cannot, therefore, be overstated.
Oral agents, which might limit the need for treatment-related infusion clinic visits and allow patients a schedule based solely on transfusion needs, have the potential to decrease rates of early discontinuation due to treatment burden and thereby result in improved quality of life, efficacy, and survival for patients with MDS. Furthermore, although most responses to HMAs have been shown to occur within 4 to 6 cycles of treatment, continuation of therapy beyond the first HI results in further improvement in response category for about half of those on treatment.7,18 Indeed, the best documented response seems to manifest at least 2 to 3 cycles after the first documented HI response, with optimal responses in 1 study manifesting as late as cycle 12 for a majority of patients.13,18
Given the ongoing burden of treatment, continued therapy on schedule must be emphasized to patients and should not be underrecognized. Early on, transfusion dependence may increase patient willingness to come to the office for treatment, but in the long term, these visits can become onerous. Ongoing studies are testing whether patients on an established parenteral HMA regimen can be switched to a lower-intensity oral regimen to maximize adherence without compromising efficacy (eg, NCT04806906). Such changes to our therapeutic approach may also improve the real-world outcomes associated with HMA treatment when compared with those seen in clinical trials. If premature discontinuation in responding patients is driven by doctors and patients unwilling to continue therapy, at least in part due to the burden of infusion clinic appointments, I believe oral therapies may help us maximize HMA benefit for patients.
CLINICAL CASE 2
A 69-year-old man with a medical history notable for obesity, diabetes, and bilateral knee osteoarthritis was referred to our practice after a preoperative CBC performed for a planned knee replacement surgery demonstrated thrombocytopenia. The CBC showed a normal WBC count, platelets of 40 000/µL, and anemia with an Hb of 10 g; the mean corpuscular volume was 106 fL. The remainder of the CBC and a comprehensive profile were normal, and nutritional workup demonstrated a sufficiency of vitamin B12, folate, and iron; ferritin was 600 (minimally elevated), and the serum erythropoietin level was 580 IU (elevated). The peripheral blood smear was notable for pelgeroid neutrophils, thrombocytopenia, and RBC anisocytosis. A BM aspirate and biopsy were hypercellular and erythroid predominant with 6% blasts and trilineage dyspoeisis. Cytogenetics were 47 XY, + 8 in 21 metaphases. NGS was notable for mutations in TET2 and ASXL1. A diagnosis of intermediate-risk MDS (IPSS score, intermediate-1; IPSS-R, 5 points [high risk]) was rendered. After discussion of prognosis and options, he elected to enroll in a phase 1b pharmacokinetic (PK) study of oral decitabine-cedazuridine fixed-dose tablet 35 mg/100 mg. Initial cycles were characterized by grade 4 thrombocytopenia and neutropenia and platelet transfusion dependence. After cycle 5 the patient had recovery of the platelet count. A BM biopsy after cycle 4 demonstrated a blast percentage of 4% with ongoing dysplasia. Referral for transplant was made, and a fully matched sibling donor was identified. The patient currently remains on protocol cycle 12 of oral decitabine-cedazuridine, given 4 days of a 28-day cycle (the dose was reduced after cycle 8 for neutropenia lasting more than 2 weeks and BM showing ongoing therapeutic response). He has declined to proceed with transplant due to concerns about transplant-related mortality. He enjoys an excellent quality of life and remains transfusion independent (TI) and nonneutropenic with a platelet count above 100 k/µL. These counts allow him to use nonsteroidal agents for management of his knee pain without bleeding risk.
PKs of parenteral vs oral HMAs
Parenteral daily HMA treatment with azacitidine and decitabine results in blood drug levels that peak between 15 and 30 minutes after administration and an elimination half-life between 1 to 2 hours for azacitidine and 35 to 40 minutes for decitabine (Table 1).31-35 The short half-life of parenterally administered HMAs is mediated largely by the ubiquitous expression of the enzyme cytidine deaminase (CDA), which is highly expressed in a variety of human tissues, including the liver, the BM, and the gastrointestinal (GI) tract. CDA rapidly degrades these agents into the inactive metabolites of uridine (Figure 1).32,36
PK characteristics of the HMAs
| Agent | Bioavailability of single oral dose (% of parenteral) | T1/2 | Tmax (range) | Cmax in ng/mL (% coefficient of variation) |
|---|---|---|---|---|
| Azacitidine IV34,40 | 100% | 4 h | 0.5 h | Similar to SQ |
| Azacitidine SQ34,40 | 89% | 4 h | 0.5 h (0.2-1.1) | 750 (54%) |
| CC-48640,52 | 11% | 0.5 h | 1 h (0.47-2) | 145 (64%) |
| Decitabine IV35 | 100% | 0.5 h | 1 h | 147 (49%) |
| Decitabine PO41 | 3.9%-14.1% | 0.36-0.93 h | 0.5 h | — |
| C-DEC43 | 60% (55-65) D1; 106% (98-114) D5 | 1.5 h | 1 h (0.3-3.0) | 145 (55%) |
| Agent | Bioavailability of single oral dose (% of parenteral) | T1/2 | Tmax (range) | Cmax in ng/mL (% coefficient of variation) |
|---|---|---|---|---|
| Azacitidine IV34,40 | 100% | 4 h | 0.5 h | Similar to SQ |
| Azacitidine SQ34,40 | 89% | 4 h | 0.5 h (0.2-1.1) | 750 (54%) |
| CC-48640,52 | 11% | 0.5 h | 1 h (0.47-2) | 145 (64%) |
| Decitabine IV35 | 100% | 0.5 h | 1 h | 147 (49%) |
| Decitabine PO41 | 3.9%-14.1% | 0.36-0.93 h | 0.5 h | — |
| C-DEC43 | 60% (55-65) D1; 106% (98-114) D5 | 1.5 h | 1 h (0.3-3.0) | 145 (55%) |
The early development of oral HMAs was characterized by the recognition that when given alone oral administration results in markedly lower peak plasma concentrations with a wide range of bioavailability when compared with IV or SQ dosing strategies.33,37-41 Table 1 provides a comparison of half-life, single-dose bioavailability (based upon area under the concentration time curve), time to maximal blood drug level (Tmax), and maximal blood concentration achieved after the administered dose (Cmax) for each drug depending upon the route of administration. Differences in bioavailability are mediated by wide inter- and intraindividual ranges of CDA expression, with higher levels in men, in those with specific single-nucleotide polymorphisms, and in the context of inflammatory drivers.32,36,42
Oral decitabine/cedazuridine
Based upon the demonstration of pharmacological equivalence to IV decitabine, in July 2020 the US regulatory authorities approved the first oral HMA, decitaine-cedazuridine (C-DEC; 35/100 mg), for patients with higher-risk MDS (intermediate/high risk by IPSS).43 In contrast with another recently approved agent, unmodified oral azacitidine (CC-486), which will be discussed subsequently, C-DEC has been shown in a randomized phase 3 study to reliably recapitulate the blood drug levels over time (cumulative area under the concentration-time curve; AUC∞; Figure 2) resulting from daily treatment for 5 days with IV decitabine, and the 2 can therefore be treated interchangeably for the purposes of clinical management.44,45
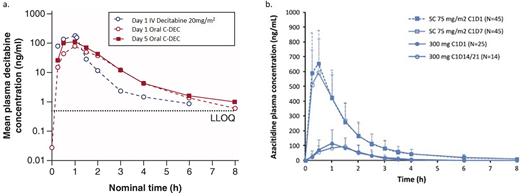
Concentration time curves for (a) IV decitabine vs oral C-DEC and (b) SQ azacitidine vs CC-486.40,46 Figure 2a was reproduced from Future Oncol. 2021;17(16):2077-2087 and was modified only by renumbering for the purpose of this article. Figure 2b was reproduced from Leukemia 2016;30(4):889-896 and was also modified only by renumbering. Both works are licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. http://creativecommons.org/licenses/by-nc-nd/4.0/
Concentration time curves for (a) IV decitabine vs oral C-DEC and (b) SQ azacitidine vs CC-486.40,46 Figure 2a was reproduced from Future Oncol. 2021;17(16):2077-2087 and was modified only by renumbering for the purpose of this article. Figure 2b was reproduced from Leukemia 2016;30(4):889-896 and was also modified only by renumbering. Both works are licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. http://creativecommons.org/licenses/by-nc-nd/4.0/
As expected, based upon the ubiquitous expression of CDA in the liver and GI tract, there is substantial first-pass metabolism of oral decitabine. Single-agent oral decitabine was shown in a phase 1 study to result in low bioavailability with substantial variation across individuals when compared with IV decitabine.41 To overcome the variable PKs resulting from differential CDA levels within and across individuals, a novel inhibitor of CDA (CDAi) given the designation E7727 (subsequently cedazuridine) was developed and extensively tested in animals.46 This agent demonstrated excellent bioavailability and a wide safety margin in nonhuman primates.46,47 In a series of early-phase studies, the combination of cedazuridine with oral decitabine (developed initially as ASTX 727 and subsequently designated decitabine- cedazuridine) was tested to develop a combination pill designed to recapitulate the PK and pharmacodynamic (PD) administration of IV decitabine dosed at 20 mg/m2 for 5 days.47 Serial blood collection was performed, and changes in global methylation using the repetitive DNA sequence long interspersed nuclear element 1 (a surrogate for global DNA methylation) was used as a tool for comparing pharmacodynamic (PD) efficacy.48 Given the substantial intra- and interindividual variability in CDA levels, the development program for this agent used each patient as their own control and compared the PK/PD for IV decitabine against the PK/PD for the oral combination.47-49 Early studies used PK/PD readouts to titrate the oral doses of E7727 and decitabine in order to create a near identity with IV decitabine administration (Figure 2a). Ultimately, the 2 were combined into a fixed-dose combination tablet containing 35 mg of decitabine and 100 mg of cedazuridine.43-45
The phase 3 study of C-DEC, upon which approval was based, randomized and treated 133 patients in a crossover design to receive either sequence A: C-DEC orally for 5 out of 28 days in cycle 1 followed by IV decitabine 20 mg/m2 × 5 out of 28 days in cycle 2 and subsequently C-DEC for cycles 3 onward or sequence B: IV decitabine 20 mg/m2 × 5 out of 28 days in cycle 1 followed by C-DEC orally daily for 5/28 days in cycle 2 and subsequently C-DEC for cycles 3 onward; cycles were to be repeated every 28 days (Figure 3). The primary end point for this phase 3 study was PK for cumulative AUC equivalence between IV and oral dosing of decitabine. Eligible patients had MDS by French-American-British classification, including chronic myelomonocytic leukemia, or MDS deemed intermediate 1, 2, or high risk by the IPSS. The clinical response to therapy was assessed according to International Working Group 2006 criteria by an independent review committee. Enrolled patients were of median age 71 years (range, 44-88), 65% were male, 12% had chronic myelomonocytic leukemia, the remainder had MDS, and 42% had >5% marrow blasts. Transfusion dependence for either RBCs or platelets was present in 43%. After a median follow-up of 12.6 months, complete remission was reported in 28 (21%) and HI in 30 (23%) patients for an overall response rate of 43%. An additional 23 patients demonstrated marrow CR without HI, although blast clearance without blood count improvement is an end point of unclear clinical significance. Adverse events (AEs) associated with C-DEC were consistent with the familiar toxicities from early cycles of parenteral HMA therapy, notably cytopenias (≥grade 3: neutropenia, 52%; thrombocytopenia, 50%; anemia, 40%; leukopenia, 21%) and infections (febrile neutropenia, 26%; pneumonia, 12%; sepsis, 7%); in contrast to CC-486, GI toxicities were not prominent.
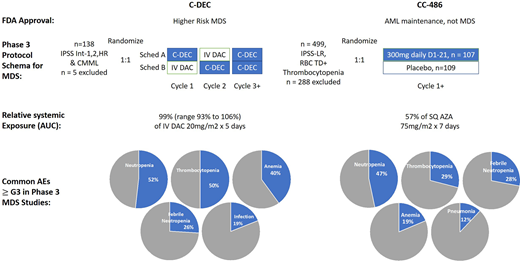
Summary comparison of the 2 completed phase 3 studies of C-DEC and CC-486 in MDS. Data abstracted from Garcia-Manero et al40,49,55 and Savona et al.45 AZA, azacitidine; DAC, decitabine.
While phase 3 data have been reported only in abstract form, they largely recapitulate the recently published results from a phase 2 multicenter randomized crossover study of similar design that enrolled and treated 80 patients. In this phase 2 study, responses (CR + HI) were observed in 44% of patients, with a majority of first responses seen by cycle 3 and best responses by cycles 5 or beyond. Among baseline transfusion-dependent (TD) patients, about 50% became TI. Overall responses and toxicity from C-DEC appear commensurate with those from parenterally administered HMAs, and this is not surprising given the identical blood drug levels (AUC) achieved with this drug. Many patients, like our patient in Case 2, will have increased transfusion needs during early cycles of therapy. The same aggressive support- ive care, including prophylactic antimicrobials (a mold-active antifungal, an oral agent that covers gram negatives, and an anti-herpes simplex virus agent) for those with prolonged neutropenia and regular evaluation of blood work for determination of transfusion needs (mandated weekly during cycles 1-3 by the clinical trial and generally performed at least 1-2 times per week in my own practice as a standard of care), particularly during early therapy cycles, is critical to optimize responses from this agent, as for all HMAs.4,24,25
Oral C-DEC is currently the only oral HMA approved for MDS. This agent was approved only for higher-risk MDS disease categories, but alternate dosing strategies with this agent are currently under investigation to determine the potential benefit for those with lower-risk disease (NCT03502668). Preliminary reports of efficacy in higher-risk MDS from the phase 2 and 3 trials demonstrate clinical activity and response that are at least comparable with (and possibly superior to) the response to IV decitabine. Although current guidelines favor SQ azacitidine as the first-choice HMA for patients with MDS given the survival benefit reported for this agent, decitabine and C-DEC are both considered acceptable alternatives.10,50 Despite this caveat, subsequent reports from clinical trials of azacitidine and decitabine suggest that survival differences in the initial clinical trials may stem from issues of trial design rather than true differences in efficacy, and a majority of practicing MDS experts would consider these agents largely interchangeable.25
Unmodified oral azacitidine
In September 2020 unmodified oral azacitidine (subsequently designated CC-486) at a dose of 300 mg daily for 14 out of 28 days (not 21 days, as tested in the MDS study) per month was approved by the US Food and Drug Administration as maintenance therapy for intermediate- and poor-cytogenetic-risk AML patients in complete remission following an intensive induction regimen; this agent is not currently approved for patients with MDS.51,52 Although this agent shares its name with parenterally administered azacitidine, the distinct PK profiles resulting from oral administration of this agent render it a different drug (Figure 2b). Substitution of CC-486 for IV or SQ azacitidine is not acceptable for patients with MDS.
As shown previously for single-agent decitabine, the extensive expression of CDA results in low bioavailability of unmodified oral azacitidine. The pilot study of CC-486 initially dosed it daily for 7 days in an attempt to recapitulate the standard parenteral dosing schedule. In this study the maximum tolerated dose was identified to be 480 mg, with a mean oral bioavailability between 6% and 20%. Relatively poor oral bioavailability could not be overcome due to dose-limiting grades 3 and 4 GI toxicity (diarrhea [12.2%], nausea [7.3%], vomiting [7.3%]) at the highest doses; other grade 3/4 AEs included febrile neutropenia (19.5%) and fatigue (9.8%).53 With this regimen, hypomethylation was lowest at day 15 and recovered to baseline by the end of the cycle.53,54 While some responses (mostly HI) were observed in this early study, decreases in methylation across a range of tested loci (the PD biomarker for activity) were less robust with CC-486 compared with the parenteral azacitidine formulation.
Given the prevailing hypothesis that the optimal hypomethylating activity of HMAs requires incorporation during S phase as well as the relatively slow proliferative rate of MDS progenitors, subsequent drug development focused on extending the dosing schedule to 14 or 21 days to allow optimal drug incorporation.38 Lower peak blood drug levels, with the sustained low-level exposure associated with this dosing strategy, would be expected to improve response rates. Indeed, extension of the dosing schedule in this manner in a PK/PD study demonstrated effective induction of hypomethylation using doses of 200 mg twice daily for 14 days or 300 mg daily for 14 or 21 days that were lowest at day 22 of treatment; hypomethylation of selected loci persisted at day 28.33 This dose schedule was tolerable, and although a majority of patients experienced GI (84%) and hematological (81%) toxicities, these were not dose limiting.38 The oral administration of CC-486 results in rapid absorption from the GI tract, with food consumption and gastric pH having limited impacts on blood drug levels.37 Subset analyses of early CC-486 studies have suggested that lower-risk patients, particularly those with thrombocytopenia, might derive particular benefit from such a low-dose oral regimen.39
Recently, a phase 3 study of CC-486 in TD lower-risk MDS patients (by IPSS criteria) reported results on 216 patients randomized 1:1 between placebo and CC-486 dosed at 300 mg daily for 21 days of a 28-day schedule (Figure 3).55 Eligibility required an Eastern Cooperative Oncology Group performance status of at least 2, thrombocytopenia (platelet count ≤75 × 109/L), and RBC transfusion dependence of at least 2 units per month for at least 2 months (International Working Group 2006 criteria).12 The primary end point for this study was RBC TI; the use of exogenous growth factors was not permitted.
Enrolled and treated patients in this study were of median age 74 years (range, 30-89), with IPSS-defined intermediate-1 risk disease (by IPSS-R, 28% had high or very high-risk MDS). They were heavily RBC TD (requiring about 3 units/month) and baseline thrombocytopenic (median 25 × 109/L; ~30% platelet TD; 40% with baseline platelet count <20 × 109/L). The study met its primary end point of RBC TI in 30% of the CC-486 patients vs 11% of placebo-treated patients; HI of the platelet count was also higher for CC-486-treated patients—24% vs 4.6%. Responses took on average 2.4 months, and decreased transfusion needs for both RBCs and platelets were sustained with CC-486 treatment (for a median of 10 months for RBCs and 12 months for platelets); bilineage improvements (in both Hb and platelet counts) were seen in about a quarter of the CC-486-treated patients. With a median follow-up of about 13 months, an underpowered overall survival estimate was the same in both arms, although the rate of AML progression was lower in the CC-486 arm. Toxicities were as expected with an HMA-based therapy and were worst in the first cycles of treatment. Most patients reported low-grade GI toxicity associated with taking CC-486. Grades 3 and 4 cytopenias (neutropenia, 47% vs 12%; thrombocytopenia, 29% vs 16%) and infections (febrile neutropenia, 28% vs 10%) were more common in the CC-486-treated patients vs those who got placebo. A higher rate of early death was reported in the CC-486 arm (16 vs 6 patients), predominantly associated with bacterial infections as well as neutropenia.
The phase 3 data reviewed above suggest substantial activity in TD lower-risk MDS patients, likely supporting an expansion of the labeled indication in the near future.55 High rates of early infectious death in this lower-risk population should at least warrant the routine use of prophylactic anti-infectives in order to mitigate risk and may give the Food and Drug Administration pause in the decision to approve this agent.24 Since CC-486 has a markedly distinct PK/PD profile from parenteral azacitidine, the two agents are not interchangeable (Figure 2b); additional studies are needed to determine whether CC-486 is appropriate for individuals with higher-risk MDS. Presently, CC-486 use in patients with MDS is limited to the context of clinical trials.
Other oral HMAs in development
Other oral HMAs are in clinical development for patients with MDS. Among these are NTX-301 (NCT04167917), a novel orally bioavailable DNMT1 inhibitor, and ASTX-030, a combination of azacitidine with oral cedazuridine (ASTX-030; NCT04256317). The latter is being developed using a combined phase 1 to 3 clinical trial based upon the PK equivalence model pioneered for C-DEC (Figure 3). It is likely that in the near future we will have approval of at least 3 oral HMAs for patients with MDS. Moreover, several studies are either planned or underway to combine oral HMAs with other promising MDS drugs, including pevonidostat, venetoclax, and sabatolimab, among others (NCT04655755, NCT04878432). Historically, combinatorial studies have failed to demonstrate improvements over the single agent.2,56 The hypotheses for combination failures include postulated inadequate dose intensity for the HMA and antagonism between agents due to convenience-dictated overlapping schedules, among other reasons.2,56 The substitution of an oral HMA in such combinations might facilitate improved dose/schedule combinations that could theoretically enhance efficacy and potentially decrease treatment modifications.2,56-58
CLINICAL CASE 3
A 78-year-old gentleman with a history of high-risk MDS (baseline hypercellular dysplastic marrow with blasts of 15%, normal karyotype in 20 metaphases, NGS with 2 distinct TET2 mutations, baseline trilineage cytopenias) has been maintained on SQ azacitidine 50 mg/m2 daily for 7 days every 28 days. The patient achieved complete remission after 6 cycles of therapy dosed at 75 mg/m2 daily for 7 days every 28 days and has been non-TD since cycle 5. Due to the development of neutropenia after cycle 9, a BM biopsy was repeated that showed ongoing CR with a hypocellular marrow. The azacitidine dose was reduced to 50 mg/m2 for 7 days of a 28-day cycle, and the patient remained TI and nonneutropenic. Due to concern about the risk from COVID-19, he requested transition to an oral regimen, having learned from a recent patient education event about the approval of C-DEC. His physician agreed to the switch, and 3 weeks into cycle 1, the patient presented with shortness of breath and fatigue. A CBC demonstrated an Hb of 5.7 g, an ANC of 0.6 × 109/µL, and platelets of 150 × 109/µL. The patient and his physician became concerned about the possibility of disease progression, and an urgent BM biopsy was performed.
Switching from parenteral to oral therapy
Recent shortages of parenteral azacitidine and concerns about the risk of COVID-19 infection posed to patients with cancer by exposure to infusion clinics have resulted in rapid substitution of C-DEC for parenteral HMAs.59,60 It is important to recognize that C-DEC results in identical PKs with IV decitabine. However, and as emphasized in the Introduction, the PKs of decitabine and azacitidine are not identical. For patients who have been maintained on dose-reduced azacitidine, the substitution of oral C-DEC represents a substantial dose increase in terms of both peak levels and total AUC exposure to HMA (Table 1). C-DEC is available only as a fixed-dose tablet; thus, dose reductions require either a decrease in the number of days of therapy or dosing on an interrupted schedule. The package insert for C-DEC suggests dose reductions from 5 to 4 days, subsequently to 3 days, and ultimately dosing every other day on days 1, 3, and 5. Due to the action of cedazuridine, which accumulates during the 5-day dosing period, the AUC for decitabine when C-DEC is dosed daily is substantially higher than when it is dosed every other day. Therefore, dose reductions to a schedule of every other day result in substantially lower decitabine exposures. When transitioning patients from an established parenteral HMA regimen, particularly when dose reductions have been made, it is important to recognize these differences in drug delivery and to either plan for up-front dose reduction to mitigate cytopenias or to reinstitute appropriate monitoring for toxicity during the initial treatment period. The patient described in case 3 underwent a BM biopsy to exclude disease progression, which demonstrated only hypocellularity. After a 2-week delay from his scheduled cycle of treatment, blood counts fully recovered. C-DEC therapy was resumed, dose reduced to 3 days per cycle this time, and blood counts were checked weekly during the next 2 cycles of therapy. No further transfusions were required, and the patient remained nonneutropenic. A failure to recognize the difference between SQ azacitidine and oral C-DEC in this case could have resulted in significant risk to this patient. Failure to plan for, or to preemptively adjust dosing in anticipation of these differences, might prompt both patient and physician reluctance to continue with oral therapy and thereby contribute to a loss of disease response or frank relapse.
Conclusions
The advent of orally bioavailable HMAs represents a substantial potential benefit for patients with MDS. While both oral decitabine (C-DEC) and oral azacitidine (CC-486) have received regulatory approval, these two agents are distinct, and only C-DEC is currently approved for patients with MDS. C-DEC fixed-dose tablets given for 5 days provide identical PK exposure to IV decitabine for 5 days, while CC-486 is dosed daily for 14 or 21 days and results in lower peak blood-drug levels than SQ or IV azacitidine but a more prolonged exposure window. The differences in PK and PD profiles for these two agents render them quite distinct in terms of both potential toxicity and mechanisms of action. As a treating clinician, I substitute C-DEC directly 1:1 for IV decitabine at a dose of 20 mg/m2 for 5 days. By contrast, CC-486 cannot be considered equivalent to IV or SQ azacitidine, and in the context of MDS, its efficacy must be tested prospectively.
Orally bioavailable HMAs have the potential to prove more convenient for MDS patients and families, sparing them repeated visits to infusion clinics for the sole purpose of drug delivery, but these agents result in substantial cytopenias that require routine assessment of transfusion needs and consideration for prophylactic antimicrobials. Most patients treated with these agents in clinical trial developed neutropenia, which was especially prominent during early cycles of therapy, and neutropenic fever was among the most frequently reported AEs. As with parenteral HMA therapy, optimal response to oral HMAs will require sustained on-schedule treatment cycles and optimized supportive care. When we consider switching to an oral regimen in an effort to optimize quality of life for a patient, particular care should be taken to recognize the different PK profiles for the new oral regimen and to anticipate the potential toxicities resulting from this change.
Acknowledgments
Elizabeth A. Griffith received research support from the Rappaport Family Foundation and the Charles D and Mary A Bauer Foundation. This work was supported by Roswell Park Comprehensive Cancer Center and National Cancer Institute grant P30CA016056.
The author would also like to acknowledge David Eifrig, Michael Nemeth, Valeria Santini, and Sophia Balderman for editorial review of the article.
Conflict-of-interest disclosure
Elizabeth A. Griffiths: research funding: Apellis Pharmaceuticals, Alexion Pharmaceuticals, Astex Pharmaceuticals, Celgene/Bristol Myers-Squibb, Celldex Therapeutics, Genentech; honoraria: Abbvie, Astex Pharmaceuticals, Boston Biomedical, Celgene/Bristol Myers-Squibb, Novartis Oncology, Takeda Oncology, Taiho Oncology.
Off-label drug use
Elizabeth A. Griffiths: This review discusses the use of CC-486/unmodified oral azacitidine for MDS, an indication that is not FDA approved.
