

Abstract
Risk stratification is crucial to the appropriate management of most cancers, but in patients with myelodysplastic syndromes (MDS), for whom expected survival can vary from a few months to more than a decade, accurate disease prognostication is especially important. Currently, patients with MDS are often grouped into higher-risk (HR) vs lower-risk (LR) disease using clinical prognostic scoring systems, but these systems have limitations. Factors such as molecular genetic information or disease characteristics not captured in the International Prognostic Scoring System–Revised (IPSS-R) can alter risk stratification and identify a subset of patients with LR-MDS who actually behave more like those with HR-MDS. This review describes the current identification and management of patients with LR-MDS whose condition is likely to behave in a less favorable manner than predicted by the IPSS-R.
Learning Objectives
Describe limitations of currently available prognostic scoring systems for patients with MDS
Discuss the current status of biologic upstaging, specifically with respect to the lower-risk patient who may require treatments usually administered to higher-risk patients
Highlight the heterogeneity in WHO-defined treatment-related MDS
Review which lower-risk patients by the IPSS-R may benefit from early consideration of allogeneic transplant
Introduction
In myelodysplastic syndromes (MDS), an optimized understanding of a patient's risk (even outside of the prognostication systems) is critical as therapeutic strategies can vary from observation of mild cytopenias or supportive treatments with transfusions through active chemotherapy and even early allogeneic hematopoietic stem transplantation (BMT). Here I consider lower-risk (LR) disease to encompass very low-, low-, and intermediate-risk categories assigned by the International Prognostic Scoring System–Revised (IPSS-R).1 The intermediate-risk group in particular is heterogeneous with a substantial proportion behaving like higher risk (HR) with early disease progression and risk of death. Treatment-related MDS (tMDS) is another prognostic challenge in the LR arena.3
CLINICAL CASE 1
A 68-year-old woman was treated 15 years ago for breast adenocarcinoma including adjuvant chemotherapy. Laboratory values now reveal a platelet (PLT) count of 102 × 109/L, hemoglobin (Hgb) of 8.2 g/dL, and white blood cell (WBC) count of 2.65 × 109/L with an absolute neutrophil count (ANC) of 0.82 × 109/L. Bone marrow biopsy specimen exhibits hypercellularity, trilineage dysplasia and 2% blasts, karyotype with 8 of 20 metaphases with chromosome 21 loss, and next- generation sequencing (NGS) revealing a RUNX1 mutation with variant allele frequency (VAF) of 26% and a monoallelic TP53 mutation with a VAF of 11%. IPSS-R score is low risk at 3 with 2 points for intermediate cytogenetics and 1 point for Hgb. She received a transfusion for anemia at diagnosis.
CLINICAL CASE 2
A 69-year-old woman also was treated for breast cancer with radiation therapy at 50 Gy. She had new anemia (Hgb, 9.9 g/dL; PLTs, 359 × 109/L; and ANC, 1.6 × 109/L). A bone marrow biopsy specimen was 30% cellular with dysgranulopoiesis and erythroid dysplasia. Blasts were 1%. Karyotype had 17 of 20 cell lines with trisomy 8 and a DNMT3A mutation with a VAF of 41%. IPSS-R score was low risk at 2, with 2 points for intermediate cytogenetics only.
Current prognostic strategies are imperfect
The use of prognostic scoring systems is common in many malignant diseases and important for predicting the survival of individuals, but these systems are often based on retrospective patient data with heterogenous treatments and often incorporate only a few salient clinical or biological features that were statistically significant in a given model. Although the IPSS and its IPSS-R1 (among others) are useful tools for clinical decision making (Table 1), these scoring systems have drawbacks and may fail to capture important prognostic information at the individual level.
Currently available prognostic scoring systems in MDS
| System | Blood | Marrow | Patient | Comments | |||||
|---|---|---|---|---|---|---|---|---|---|
| Hgb | PLTs | ANC | Blasts | Cyto | Age | Txn | PS | ||
| IPSS | + | + | + | + | + | + | Has been revised now (2012) using the same prognostic parameters (number and depth or cytopenias, marrow blast percentage [more granular] and karyotype), was able to reclassify nearly 25% of lower-risk MDS patients into a higher-risk category | ||
| WPSS | + | + | + | Time dependent which is useful over the course of patient's disease | |||||
| MDS LR | + | + | + | + | + | + | May be applied specifically to lower-risk disease with more variables | ||
| MDAPSS | + | + | + | + | + | + | + | Contains more clinical variables, especially age and transfusion burden, which could upstage a patient | |
| FPSS | + | + | + | Higher-risk disease and includes azacytidine-treated patients, likely is not useful in lower-risk patients, even for upstaging | |||||
| IPSS-R | + | + | + | + | + | + | Used most commonly at diagnosis and for clinical trial eligibility, limitations discussed in text | ||
| System | Blood | Marrow | Patient | Comments | |||||
|---|---|---|---|---|---|---|---|---|---|
| Hgb | PLTs | ANC | Blasts | Cyto | Age | Txn | PS | ||
| IPSS | + | + | + | + | + | + | Has been revised now (2012) using the same prognostic parameters (number and depth or cytopenias, marrow blast percentage [more granular] and karyotype), was able to reclassify nearly 25% of lower-risk MDS patients into a higher-risk category | ||
| WPSS | + | + | + | Time dependent which is useful over the course of patient's disease | |||||
| MDS LR | + | + | + | + | + | + | May be applied specifically to lower-risk disease with more variables | ||
| MDAPSS | + | + | + | + | + | + | + | Contains more clinical variables, especially age and transfusion burden, which could upstage a patient | |
| FPSS | + | + | + | Higher-risk disease and includes azacytidine-treated patients, likely is not useful in lower-risk patients, even for upstaging | |||||
| IPSS-R | + | + | + | + | + | + | Used most commonly at diagnosis and for clinical trial eligibility, limitations discussed in text | ||
The prognostic scoring systems are listed in order of publication with annotations on the variables included and comments on their use in lower risk disease.
Cyto, cytogenetics; FPSS, French Prognostic Scoring System; MDAPSS, MD Anderson Prognostic Scoring System; MDS LR, scoring system for patients with lower-risk MDS; PS, performance status; Txn, transfusions; WPSS, WHO Classification-Based Prognostic Scoring System.
The key limitation to the prognostication systems is their inability to capture all relevant biology. For example, cytogenetics is the only biological parameter included in the IPSS-R, and unfortunately, although an IPSS revision incorporating NGS molecular data is in development, this has yet to be reported. At the 2015 meeting of the American Society of Hematology, Bejar et al2 from the International Working Group for Prognosis in MDS–Molecular Committee reviewed analyses that showed somatic mutations in MDS are associated with clinical features and predict prognosis independent of the IPSS-R. Bersanelli et al4 recently showed that integration of clinical data with NGS profiling improves the accuracy of currently available prognostic scores. The authors provided evidence from another large, international database that MDS could be classified into 8 distinct subtypes according to specific genomic features.4 These subgroups do not correlate with morphologic categories defined by the current World Health Organization (WHO) classification and displayed significantly different clinical phenotypes and outcome.4
The IPSS-R was based only on standard G-banded metaphase karyotyping. A recent study examined whole-genome sequencing (WGS) of patients with MDS or acute myeloid leukemia (AML) for feasibility assessment.5 Among 42 patients with MDS in this study, the conventional results (translocations and copy number variation) were confirmed in all cases by WGS; nearly 25% had additional chromosomal abnormalities identified by WGS that, if incorporated into IPSS-R, would have altered the risk category.5 Although I do know that a chromosomal alteration identified only by WGS has the same prognostic meaning as that identified by metaphase karyotyping, I believe this suggests further need for efficient incorporation of additional cytogenetic and molecular data into prognostic scoring systems.
CLINICAL CASES 2 (continued)
Patient 1 went on to receive 14 cycles of azacytidine. Transplant options were pursued but no available donor located. She remained red blood cell transfusion dependent (RBC-TD) with Hgb routinely below 8 g. Repeat marrow assessment continued to show LR features; therapy was not altered. She ultimately progressed to AML 19 months after presentation. Patient 2 has been followed with every 3-monthly blood counts and biennial marrow assessments for 6 years with near-complete stability.
Patients 1 and 2 highlight further significant clinical limitation in the IPSS-R: how to categorize tMDS. About 15% of MDS are tMDS and generally considered at HR. These disorders are of clinical importance for patients and of growing academic interest, especially as patients live longer due to more effective therapies. With closer follow-up, this category is likely to be the largest increase in patients with “higher-risk” low-risk MDS. The IPSS-R cohort did not contain any patients with tMDS, and the prognosis of these patients is complicated by several non-MDS factors, including prior toxicities of therapy (transfusions, alloantibodies, organ limitations), as well as other medical comorbidities. There is the common perception that patients with tMDS have high-risk disease, regardless of IPSS-R score, and therefore warrant aggressive therapy. Patient 2's course would argue against this. According to the current definition of the WHO, tMDS is defined by the history of receipt of chemotherapy and/or radiotherapy for nonmyeloid malignancy or medical condition.6 This cannot always allow for disease that may not truly be attributed to the previous therapy (perhaps in patient 2) as the disease behavior is more akin to de novo MDS. Nonetheless, the IPPS-R has been shown to be prognostically inadequate in tMDS, as in patient 1.7 The US MDS Clinical research consortium reported shorter survival in tMDS than in de novo MDS, including in LR patients.8 Zeidan et al8 analyzed outcomes in 370 patients with tMDS to understand the prognostic utility of current risk stratification tools in tMDS. The overall survival (OS) of patients with tMDS was significantly worse than those with de novo disease (median OS, 19 vs 46 months).
The genetics of tMDS has been investigated to explain if their “risk heterogeneity” (as displayed by both cases) might be partially related to inadvertent inclusion of coincidental second disease due to genetic predisposition.9 A history of therapy might mean there was clonal selection at some ancestral point, but the biology is heterogeneous, and some have more indolent disease, as in patient 2. Previous cancer therapy with radiation, platinum, and topoisomerase II inhibitors, as in patient 1, preferentially selects for mutations in DNA damage response genes (TP53, PPM1D, CHEK2) in a recent large series.10 These latter patients will likely all behave in an HR fashion, regardless of IPSS-R, and should be treated more aggressively with serially reassessment and alterations to therapeutics. The management of patients with tMDS remains challenging, and careful surveillance and openness to change therapy as course dictates—to lower intensity or higher intensity—are critical.
CLINICAL CASE 3
A previously healthy 39-year-old man sought treatment for fatigue and cytopenias. He had no splenomegaly. His total WBC count was 2.4 × 109/L, ANC was 1.36 × 109/L, Hgb was 7.9 g/dL, and PLTs were 99 × 109/L. Marrow biopsy specimen was 95% cellular with erythroid predominance, trilineage dyspoiesis, and 3% blasts. There were 25% to 30% ring sideroblasts with a diffuse increase in reticular fibrosis. His final diagnosis was MDS with ring sideroblasts and multilineage dysplasia. His karyotype was 46,XY, and NGS showed SF3B1 R625H with a VAF of 38% without comutations to suggest myelofibrosis. IPSS-R score was intermediate-risk disease at 3.5 with 1 for cytogenetics, 1 for 3% blasts, 1 for Hgb less than 8 g, and 0.5 for PLTs less than 100 × 109/L.
Clinical limitations in use of prognostic scoring systems also exist. Table 2 summarizes additional potential poor prognostic factors in LR patients that fall outside the IPSS-R. Grade 2 or more marrow fibrosis may worsen prognosis of patients with MDS.11 RBC transfusion dependency is an independent poor prognosticator. Recently, Hiwase et al12 examined whether RBC-TD adds prognostic value to the IPSS-R. In a multivariate analysis, their cohort demonstrates that development of RBC-TD at any time during the course of MDS is associated with poor OS, independent of IPSS-R.12 The LeukaemiaNet MDS registry recently demonstrated that a relative PLT drop of more than 25% results in a 22% OS at 5 years.13 When the PLT drop is combined with RBC-TD at 6 months from diagnosis, the OS is a staggeringly low 9%.13 This is a straightforward and noninvasive way to predict evolution to HR disease for LR MDS. The addition of the intermediate-risk group to the IPSS-R has also been a clinical challenge as it is most often considered still LR yet is highly heterogeneous. Benton et al14 performed an analysis of 298 intermediate-risk patients to assess HR features. Age older than 66 years, peripheral blood blasts of 2% or more, and RBC transfusion were significantly associated with inferior survival.14 Patient 3 highlights several of these issues.
Features in lower-risk MDS that suggest higher-risk behavior
| MDS characteristic | Feature associated with lesser prognosis |
|---|---|
| Etiology of MDS | Treatment related, can behave in a heterogenous fashion |
| Fibrosis in core biopsy | Grade 2 or higher |
| Cytopenia | Symptomatic neutropenia |
| Decrease in PLTs >25% | |
| Ongoing RBC transfusion dependence | |
| Anemia or thrombocytopenia refractory to transfusions | |
| Karyotype | Clonal emergence of unfavorable karyotype |
| Somatic mutations | Multiple mutations (≥3 somatic mutations) |
| T53, RUNX1, ASXL1 mutations | |
| Absence of SF3B1 mutation (especially in MDS with ring sideroblasts) | |
| Multiple somatic mutations | |
| Inherited predisposition | Patients with known germline variant in their disease may be less likely to respond to traditional therapies and require stem cell therapy sooner |
| Treatment response | Primary treatment failure vs secondary failure |
| MDS characteristic | Feature associated with lesser prognosis |
|---|---|
| Etiology of MDS | Treatment related, can behave in a heterogenous fashion |
| Fibrosis in core biopsy | Grade 2 or higher |
| Cytopenia | Symptomatic neutropenia |
| Decrease in PLTs >25% | |
| Ongoing RBC transfusion dependence | |
| Anemia or thrombocytopenia refractory to transfusions | |
| Karyotype | Clonal emergence of unfavorable karyotype |
| Somatic mutations | Multiple mutations (≥3 somatic mutations) |
| T53, RUNX1, ASXL1 mutations | |
| Absence of SF3B1 mutation (especially in MDS with ring sideroblasts) | |
| Multiple somatic mutations | |
| Inherited predisposition | Patients with known germline variant in their disease may be less likely to respond to traditional therapies and require stem cell therapy sooner |
| Treatment response | Primary treatment failure vs secondary failure |
Adverse molecular features may “upstage” an LR patient
Most cases of MDS have 1 or several somatic gene mutations. SF3B1MUT is generally considered a favorable prognostic marker.15 Despite this, SF3B1MUT MDS is heterogeneous in its pathologic features, treatment responses, and clinical outcomes,16 as in patient 3. Understanding this heterogeneity is crucial for disease prognostication and management, and it is relevant to a recent proposal by the MDS International Working Group that SF3B1MUT MDS should be classified as a distinct nosologic entity.15 Although historically, SF3B1MUT generally represents indolent disease in LR MDS, comutation with RUNX1 and EZH2 has been associated with a worse prognosis.15
The independent poor prognostic value of many somatic mutations, including ASXL1, SRSF2, RUNX1, and TP53, has been established retrospectively.17 The integration of these unfavorable mutations in specific scoring systems for LR MDS has been reported. In addition, an increased number of mutations present in a single patient also suggests a less favorable outcome.18
Of particular importance is the TP53 mutation. Specifically, this is present in 20% of cases of LR MDS with isolated deletion 5q minus syndrome (del5q; but rare in other LR MDS) and associated with an HR of AML and poorer survival.19 However, a recent study analyzed more than 3000 patients with MDS for TP53 mutations and allelic imbalances.19 One-third of TP53-mutated patients had monoallelic mutations, whereas two-thirds had multiple mutations consistent with biallelic targeting. Established associations with complex karyotype, few co-occurring mutations, high-risk presentation, and poor outcomes were specific to multihit patients only. Interestingly, monoallelic patients did not differ from TP53 wild-type patients in outcomes and response to therapy in this series.19 Nonetheless, most still use the presence of TP53 mutation (even monoallelic) to upstage LR disease given its long association with adverse outcomes, as in patient 1.
CLINICAL CASE 3 (Continued)
Four months after diagnosis, the patient remained well, but blood counts were lower and treatment warranted for cytopenias, despite still intermediate risk by IPSS-R on repeat marrow. Concern for his MDS diagnosis at age less than 40 years prompted deeper genetic assessment. Ultimately, a germline predisposition gene associated with myeloid malignancies was identified in the patient (and also in his brother and father) with the RTEL1 mutation. He then underwent unrelated donor BMT with normal counts and full donor chimerism without complications at 1 year.
Inherited predisposition to MDS may alter risk assessment
Inherited variants are recognized increasingly as predisposing patients to MDS, and germline susceptibility to myeloid malignancies is included in the latest WHO classification of hematopoietic malignancies. Moreover, clinical guidelines now call for assessment of germline predisposition at MDS diagnosis.20,21 A recent series showed that the frequency of germline variants in adults aged 18 to 40 years diagnosed with MDS is high (consistent with other reports), and the associated germline syndrome is often not apparent from the patient's medical or family history.22 Patient 3, with his younger age, illustrates this need for deeper genetics assessment. These patients may behave in a variety of ways with HR manifestations; coexisting somatic mutations are not uncommon in these patients.23 There are also many benefits of real-time differentiation to inherited predisposition beyond potential biologic understanding of increased risk features to LR disease.20
Outcomes in LR disease with currently available therapies
Figure 1 depicts many of the standard treatments24-27 often employed in LR disease, regardless of HR features, as well as frequent need for reassessment if response to treatment is not positive. Although hypomethylating agents (HMAs), such as azacitidine and decitabine, and lenalidomide (LEN) are not approved in many countries for LR disease, the focus here is on drugs for consideration in our patients and the available data in the literature. For thrombocytopenic LR patients, eltrombopag is modestly clinically effective in raising PLT counts and reducing bleeding events.28 It is not currently approved for MDS and not yet known if it will be safe. The assessment of long-term safety and efficacy of eltrombopag and its effect on survival (phase 2 part of study) is still ongoing28 and likely will be held to a high standard to ensure no increased clonal evolution. Romiplostim has also been studied in MDS and reduces PLT transfusions and bleeding events with good OS without increasing rates of AML.29 Clinical trials of novel therapies in LR disease, such as imetelstat,30 may yet be able to modify disease risk and change the natural history.
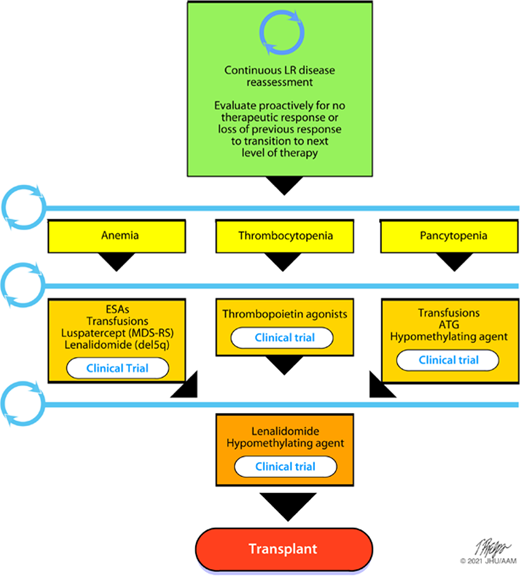
Therapeutic paradigm in LR MDS. MDS-RS, myelodysplastic syndrome with ring sideroblasts.
Therapeutic paradigm in LR MDS. MDS-RS, myelodysplastic syndrome with ring sideroblasts.
Prebet et al. have looked at outcomes posttreatment with LEN in LR MDS with and without del5q.31,32 In patients with del5q, OS following LEN failure was a mere 23 months,31 whereas in non-del5q, it was 43 months.32 LEN resistance in del5q is also associated with a high risk of AML and a 5-year probability of survival of only 25%.31 For non-del5q, subsequent therapy with HMAs was associated with a prolonged survival compared with best supportive care (median OS, 51 vs 36 months, P = .01).32 This suggests an early switch to HMAs in LEN-resistant patients is reasonable. The time to resistance or failure of therapy varies in the literature, but a trial probably should be no longer than 16 weeks for non-del5q patients as more than 90% of responders were seen in the phase 3 MDS-005 trial after those 4 cycles.25 In the same study, the authors explored gene mutations and response to LEN.33 The analyses revealed that patients with 4 or more mutations were more likely to have a high serum erythropoietin (EPO) level (>500 mU/mL); in addition, high serum EPO level was associated with the presence of mutations in any of 5 genes associated with poorer outcomes in the Bejar et al34 analysis (ASXL1, ETV6, EZH2, RUNX1, and TP53). Taken together with the observed association between ASXL1 mutations and low response rate to LEN, this provides further insight into previous data that high baseline EPO level predicts for nonresponse to LEN in these LR patients.25,33 Finally, even in LR patients, nonresponse to HMAs was associated with a median survival of 17 months in a US MDS consortium study,35 which correlates with increasing number of mutations.36 Thus, response to all lines of therapy in LR MDS should also be considered for transitions of therapy. Clinical trials should also always be considered. Increasingly, trials for patients with MDS include a clause for eligibility that allows for upstaging, such as “patients with intermediate risk by R-IPSS with high-risk molecular features including TP53, ASXL1, EZH2, and/or RUNX1 mutations are also eligible.” I believe this is a reasonable practice to apply broadly.
Consideration of allogeneic transplantation (BMT) in LR disease
Historically, BMT has been reserved for patients with HR MDS, given its potential curative benefit is offset by the morbidity and mortality that can result from the procedure.37,38 Nonetheless, efforts have been made to weigh the toxicities of this “higher risk, higher reward” procedure in LR MDS, and many LR patients are offered BMT. The National Comprehensive Network Guidelines and other consensus guidelines recommend BMT for patients with MDS early in their disease if they have intermediate 2 or high-risk category per the IPSS and later during their disease course (prior to AML progression) if they have LR disease.
Although no prospective series exist currently, there are reported outcomes of BMT in LR patients included in many retrospective series.39-43 Only 1 dedicated retrospective study reviewed outcomes in LR patients, and the results were not encouraging.42 In a study of patients who underwent transplantation between 2000 and 2011 by the European Society for Blood and Marrow Transplantation (EBMT), 246 IPSS low-risk (21%) or intermediate 1 (79%) patients had a 3-year OS and PFS of 57% and 54%, respectively. There was a high nonrelapse mortality at 30%. The best outcome was seen in patients who received peripheral blood stem cells and had a matched related donor, although alternative donors were relatively rare. The conditioning regimen intensity and patient age did not appear to influence outcome in this heterogenous series.42 In a series from Asia, LR patients had a significantly better outcome than HR patients.43 Prognosis was also significantly influenced by the genetic risk. LR patients who display HR features such as a complex karyotype or TP53 mutations do poorly, whereas LR 5q minus patients had an OS of about 40%.43 Additional explanations of incorporation of biologic data into BMT outcome analyses are seen in a large series from the Center for International Blood and Marrow Transplant Research. The authors reported 1514 patients with MDS who underwent transplantation, of whom 116 were considered LR. These patients were considered favorable, with an age at transplant from less than 1 year to less than 40 years, no tMDS, no TP53 or Ras pathway gene mutation, no thrombocytopenia, and marrow blasts less than 15%.40 Although not a classical definition of LR patients, this cohort of children and young adults had a favorable 3-year OS of 82%.40 The supplementary data review OS curves by IPSS specifically and suggest that low-risk and intermediate 1 patients without TP53 mutations have an OS at 3 years of approximately 50%.
To aid in decision making, some series from the EBMT and Center for International Blood and Marrow Transplant Research have developed scoring systems or decision models that adjust to individual patient risk. Although not specific to LR patients, the molecular and other clinical features associated with less desirable outcomes can been seen. Both of these series had an OS in LR patients of about 70% at 3 years or 4 years.41,44 The Gruppo Italiano Trapianto Midollo Osseo e Terapia Cellulare reported a BMT Markov analysis on 1728 patients with MDS categorized by the IPSS-R. Similar to older analyses, a survival advantage was seen when delaying transplant in very low- and low-risk patients, whereas postponement was deleterious in intermediate-risk patients.45 This is in keeping with the current approach to only consider BMT in LR patients with disease evolution or lack of response to primary treatments. Patients with increased RBC needs, lack of response to HMAs, bone marrow fibrosis, HR molecular features, and tMDS are associated with a poor prognosis, and early consideration for BMT is often suggested.35,46,47 Genomic features are relevant for predicting survival after BMT, supporting the rationale to include this information for transplantation decision making in MDS.48,49 Many of these features would have been applicable in both patients 1 and 3.
Additional data including prospective studies, potentially with a randomized design compared with the current standard of delaying BMT, and including a wider donor pool with haploidentical donors are needed for an improved understanding and establishment of the BMT approach for LR patients. An ongoing prospective nonrandomized trial in France is enrolling patients with LR disease and at least 1 high-risk feature (NCT02757989), and results will be useful to the field. Nonetheless, BMT remains the only potentially curative option for MDS, and no options available at present seem to be able to replace this path to potential cure; we must use this for patients when the risk/benefit profile is rational.
Conclusions
LR MDS is a heterogeneous group of disorders. Currently available prognostic scoring systems aid in risk stratification but do not capture all important variables, and serial reassessment of the patient to fully classify individual risk is important. The goal is a proactive approach in all LR patients to extend quantity of life with quality through use of all available treatments available.
Conflict-of-interest disclosure
Amy E. DeZern has received honoraria from Bristol Meyers Squibb, Novartis, Taiho, and Abbvie as a consultant.
Off-label drug use
Amy E. DeZern: imetelstat or eltrombopag for MDS.
