

Abstract
At least 16 genetically determined conditions qualify as red blood cell enzymopathies. They range in frequency from ultrarare to rare, with the exception of glucose-6-phosphate dehydrogenase deficiency, which is very common. Nearly all these enzymopathies manifest as chronic hemolytic anemias, with an onset often in the neonatal period. The diagnosis can be quite easy, such as when a child presents with dark urine after eating fava beans, or it can be quite difficult, such as when an adult presents with mild anemia and gallstones. In general, 4 steps are recommended: (1) recognizing chronic hemolytic anemia; (2) excluding acquired causes; (3) excluding hemoglobinopathies and membranopathies; (4) pinpointing which red blood cell enzyme is deficient. Step 4 requires 1 or many enzyme assays; alternatively, DNA testing against an appropriate gene panel can combine steps 3 and 4. Most patients with a red blood cell enzymopathy can be managed by good supportive care, including blood transfusion, iron chelation when necessary, and splenectomy in selected cases; however, some patients have serious extraerythrocytic manifestations that are difficult to manage. In the absence of these, red blood cell enzymopathies are in principle amenable to hematopoietic stem cell transplantation and gene therapy/gene editing.
Learning Objectives
Update clinical, hematologic, biochemical, and molecular approaches to the diagnosis of red blood cell enzymopathies
Pinpoint the role of supportive treatment, targeted treatment, and advanced forms of treatment in the management of red blood cell enzymopathies
Update knowledge about the molecular basis of red blood cell enzymopathies
Introduction
Red blood cell enzymopathies are genetic disorders affecting the intraerythrocytic metabolism.1,2 Red blood cells, having sacrificed their nucleus and other organelles to the unselfish task of providing oxygen to all other cells, are limited in their capacity to withstand metabolic impairment and also limited in how they will be affected: when one important enzyme is deficient, the red blood cell life span is nearly always compromised, with consequent hemolysis. Most red blood cell enzymes are ubiquitously expressed “housekeeping” enzymes (Table 1); therefore, there may be pathology in other tissues as well. Because red blood cells do not have protein synthesis, however, they are in general more vulnerable.
List of red blood cell enzymopathies underlying CNSHA*
| Enzyme (Acronym) | Gene | Chromosomal location | Extraerythrocytic Clinical Manifestations† | Notes | |
|---|---|---|---|---|---|
| Glycolytic pathway36 | Hexokinase (HK) | HK1 | 10q22 | May benefit from splenectomy; bone marrow transplantation (BMT) has been done. | |
| Glucose-6-phosphate isomerase (GPI) | GPI | 19q31.1 | Rarely myopathy, central nervous system (CNS) | Ranks second in frequency within the glycolytic pathway group (after PK). May benefit from splenectomy. | |
| Phosphofructokinase (PFK) | PFKM | 12q13.3 | Myopathy, myoglobinuria | Primarily a muscle disease, with glycogenosis in muscle. CNSHA mild.7 | |
| Aldolase | ALDOA | 16q22-24 | Myopathy, CNS | Fever may trigger potentially fatal massive rhabdomyolysis.37 | |
| Triose phosphate isomerase (TPI) | TPI1 | 12p13.31 | CNS (severe), cardiomyopathy | Disease very rare, but same mutation (p. Glu104Asp) found in several cases, suggesting founder effect. | |
| Glyceraldehyde-3- phosphate dehydrogenase (GAPD) | GAPDH | 12p13.31- | Myopathy | Hemolytic anemia reported in some cases but link with enzyme deficiency not clearly established. | |
| Bisphosphoglycerate mutase (DPGM) | BPGM | 7q33 | No hemolysis; erythrocytosis can be attributed to low 2,3-DPG, left-shifted Hb-O2 dissociation curve, relative hypoxia. | ||
| Phosphoglycerate kinase (PGK) | PGK1 | Xq21.1 | CNS myopathy | May benefit from splenectomy; BMT has been done. Early-onset parkinsonism reported.38 | |
| Pyruvate kinase (PK) | PKLR | 1q22 | Iron overload | Ranks first in frequency in the glycolytic enzyme group. May benefit from splenectomy; BMT has been done. | |
| Redox16,39 | Glucose-6-phosphate dehydrogenase (G6PD) | G6PD | Xq28 | Very rarely granulocytes | In almost all cases, only AHA from exogenous trigger‡; CNSHA only with class I variants. |
| Glutathione synthase | GSS | 20q11.22 | CNS | May be associated with high 5-oxoproline and metabolic acidosis.40 | |
| Glutathione reductase | GSR | 8p12 | Cataracts | AHA from exogenous trigger (favism). | |
| γ-glutamylcysteine synthase | GCLC | 6p12.1 | CNS | Mutations affect catalytic subunit.41 | |
| Cytochrome b5 reductase (CBR) | CYB5R3 | 22q13.2 | CNS | Methemoglobinemia rather than hemolysis. | |
| Nucleotide metabolism | Adenylate kinase (AK) | AK1 | 9q34.11 | CNS | May benefit from splenectomy. |
| Pyrimidine 5′-nucleotidase (P5N) | NTSC3A | 7p14.3 | Ranks third in frequency among all red blood cell enzymopathies (leaving aside G6PD). May benefit from splenectomy. |
| Enzyme (Acronym) | Gene | Chromosomal location | Extraerythrocytic Clinical Manifestations† | Notes | |
|---|---|---|---|---|---|
| Glycolytic pathway36 | Hexokinase (HK) | HK1 | 10q22 | May benefit from splenectomy; bone marrow transplantation (BMT) has been done. | |
| Glucose-6-phosphate isomerase (GPI) | GPI | 19q31.1 | Rarely myopathy, central nervous system (CNS) | Ranks second in frequency within the glycolytic pathway group (after PK). May benefit from splenectomy. | |
| Phosphofructokinase (PFK) | PFKM | 12q13.3 | Myopathy, myoglobinuria | Primarily a muscle disease, with glycogenosis in muscle. CNSHA mild.7 | |
| Aldolase | ALDOA | 16q22-24 | Myopathy, CNS | Fever may trigger potentially fatal massive rhabdomyolysis.37 | |
| Triose phosphate isomerase (TPI) | TPI1 | 12p13.31 | CNS (severe), cardiomyopathy | Disease very rare, but same mutation (p. Glu104Asp) found in several cases, suggesting founder effect. | |
| Glyceraldehyde-3- phosphate dehydrogenase (GAPD) | GAPDH | 12p13.31- | Myopathy | Hemolytic anemia reported in some cases but link with enzyme deficiency not clearly established. | |
| Bisphosphoglycerate mutase (DPGM) | BPGM | 7q33 | No hemolysis; erythrocytosis can be attributed to low 2,3-DPG, left-shifted Hb-O2 dissociation curve, relative hypoxia. | ||
| Phosphoglycerate kinase (PGK) | PGK1 | Xq21.1 | CNS myopathy | May benefit from splenectomy; BMT has been done. Early-onset parkinsonism reported.38 | |
| Pyruvate kinase (PK) | PKLR | 1q22 | Iron overload | Ranks first in frequency in the glycolytic enzyme group. May benefit from splenectomy; BMT has been done. | |
| Redox16,39 | Glucose-6-phosphate dehydrogenase (G6PD) | G6PD | Xq28 | Very rarely granulocytes | In almost all cases, only AHA from exogenous trigger‡; CNSHA only with class I variants. |
| Glutathione synthase | GSS | 20q11.22 | CNS | May be associated with high 5-oxoproline and metabolic acidosis.40 | |
| Glutathione reductase | GSR | 8p12 | Cataracts | AHA from exogenous trigger (favism). | |
| γ-glutamylcysteine synthase | GCLC | 6p12.1 | CNS | Mutations affect catalytic subunit.41 | |
| Cytochrome b5 reductase (CBR) | CYB5R3 | 22q13.2 | CNS | Methemoglobinemia rather than hemolysis. | |
| Nucleotide metabolism | Adenylate kinase (AK) | AK1 | 9q34.11 | CNS | May benefit from splenectomy. |
| Pyrimidine 5′-nucleotidase (P5N) | NTSC3A | 7p14.3 | Ranks third in frequency among all red blood cell enzymopathies (leaving aside G6PD). May benefit from splenectomy. |
The molecular basis of an enzymopathy is in the mutation(s) in the respective gene. Most enzymopathies are autosomal recessive disorders. Therefore, the patient has a mutation in each of the 2 allelic genes encoding the respective enzyme: the mutation may be the same in both alleles (homozygosity), or there may be 2 different mutations (compound heterozygosity or biallelic mutations). In most cases the mutation causes instability of the protein: since mature red blood cells cannot make proteins, the enzyme activity with which a reticulocyte is endowed will decay as the red blood cell ages in circulation.
Since we are dealing with ubiquitously expressed genes, the presence of such manifestations is not surprising. Whether they are present or not, and to what extent, depends mainly on 3 factors: (1) nonerythroid cells may express alternative forms of a particular enzyme, from the same gene or from other genes; (2) if the main mechanism of deficiency is enzyme instability, cells other than erythrocytes may compensate by increased biosynthesis; (3) if the main mechanism of deficiency is a qualitative change (eg, in the active site), all cells expressing the gene will be affected. The last 2 factors may explain why extraerythrocytic manifestations may vary even within the same enzymopathy.
G6PD deficiency is widespread in malaria-endemic areas, consequent on malaria having been the selective agent for this polymorphism.42 In areas endemic for Plasmodium vivax malaria, a test for G6PD deficiency should be carried out before giving primaquine or tafenoquine, the only drugs that prevent relapse. This has been a strong stimulus for the development of quantitative point-of-care tests for G6PD deficiency.43
The main metabolic fuel of the red blood cell is plasma glucose, and its breakdown within the red blood cell provides 2 key compounds: adenosine triphosphate (ATP), required mainly for the operation of the cation pump, and nicotinamide adenine dinucleotide phosphate, reduced (NADPH), required mainly for keeping up the supply of reduced glutathione (GSH) essential for the detoxification of hydrogen peroxide (H2O2) and of other reactive oxygen species (ROS). ATP is produced by reactions of the “classic” glycolytic pathway; NADPH is produced by the pentose phosphate pathway (Figure 1).3 Therefore, it is natural to classify red blood cell enzymopathies according to these 2 groups. A third group comprises enzymes involved in nucleotide metabolism (Table 1).
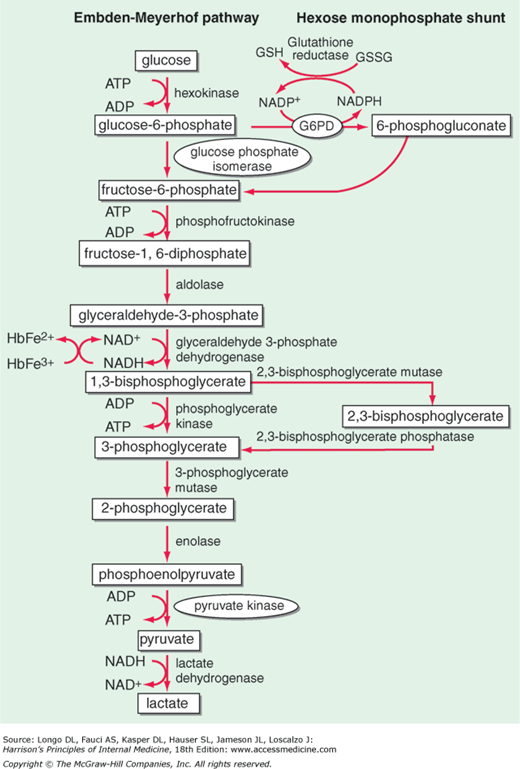
Red blood cell metabolism. Glycolysis (the Embden-Meyerhof pathway) generates ATP required for cation transport and for membrane maintenance, while NADH maintains hemoglobin iron in a reduced state. The hexose monophosphate shunt generates the NADPH that is used to reduce GSH, which protects the red blood cell against oxidant stress (Figure 7); 6-phosphogluconate, after decarboxylation, can be recycled via pentose sugars to glycolysis. At the side of glycolysis is the Rapoport-Luebering cycle, which provides 2,3-isphosphoglycerate (2,3-DPG): its level is a critical determinant of the oxygen affinity of hemoglobin. G6PD deficiency is highly prevalent in many parts of the world (Figure 6); all other enzymopathies are rare to ultrarare. Among them, the order of prevalence is PK, followed by P5N, followed by GPI.
Red blood cell metabolism. Glycolysis (the Embden-Meyerhof pathway) generates ATP required for cation transport and for membrane maintenance, while NADH maintains hemoglobin iron in a reduced state. The hexose monophosphate shunt generates the NADPH that is used to reduce GSH, which protects the red blood cell against oxidant stress (Figure 7); 6-phosphogluconate, after decarboxylation, can be recycled via pentose sugars to glycolysis. At the side of glycolysis is the Rapoport-Luebering cycle, which provides 2,3-isphosphoglycerate (2,3-DPG): its level is a critical determinant of the oxygen affinity of hemoglobin. G6PD deficiency is highly prevalent in many parts of the world (Figure 6); all other enzymopathies are rare to ultrarare. Among them, the order of prevalence is PK, followed by P5N, followed by GPI.
Features of hemolytic anemias due to enzymopathies can be best illustrated through clinical summaries of individual patients.
CLINICAL CASE
A 1-year-old boy born in 1963 from parents not known to be consanguineous, although both were from Morcone (a small town not far from Naples, Italy), was found by his local pediatrician to have anemia with reticulocytosis. Since birth he had had moderate jaundice that did not respond to barbiturates. Over several years his hemoglobin (Hb) level fluctuated in the range from 7 to 11 g/dL, and he received several blood transfusions. At the age of 18, when he was fully reassessed (B. Rotoli, MD, in Naples), physical examination showed pallor, mild icterus, and splenomegaly. There were no neurological signs or muscle weakness. Hb was 10.4 G/dL; mean corpuscular volume, 104 fL, reticulocytes, 480 × 109/L; and unconjugated bilirubin, 5 mg/dL; erythrocyte morphology showed anisocytosis and basophilic stippling in 4% of cells. There were no abnormal hemoglobins, osmotic fragility was normal, and the autohemolysis test showed increased lysis not corrected by glucose. A 51 Cr study showed reduction of the red blood cell half-life by about 40%, with increased radioactive uptake in both the spleen and liver. A panel of red blood cell enzyme assays (A. Zanella, MD, Milan) revealed a marked deficiency of glucose-6-phosphate isomerase (GPI).4 The patient, who had been placed on daily folic acid (5 mg/day), continued to require an average of 2.5 blood units per year until, at the age of 25, he developed obstructive jaundice due to gallstones. He underwent surgery that included cholecystectomy, choledochoduodenostomy, and splenectomy. The spleen weighed 500 g; microscopic examination showed marked congestion, erythrophagocytosis, and small foci of erythropoiesis. A liver biopsy showed evidence of cholestasis. The Hb level increased and remained for years in the range of 10 to 11 g/dL; therefore, regular blood transfusions were no longer needed. Reticulocytes remained high (in the range of 250-350 × 109/L). At the age of 27, the patient completed his doctorate in philosophy. When the patient was 31 years old, sequencing of the GPI gene revealed that the patient was homozygous for a p.Q343R mutation identical to that independently observed at about the same time in a patient from Japan.5,6
At the age of 44, the patient developed severe jaundice that persisted for weeks (the bilirubin peaked at 80 mg/dL) and was admitted to a unit that specialized in liver transplantation (Dr G. Ramadori, Göttingen). Imaging studies revealed an accessory spleen. A liver biopsy (reviewed by Dr Tania Roskam, Leuven) showed massive bilirubin overload, cholestasis, ballooned hepatocytes, ceroidosis of the Kupffer cells, and moderate centrolobular perisinusoidal fibrosis but no evidence of cirrhosis. On supportive treatment (including transfusion of 10 units of blood), the patient gradually improved. Quantitative magnetic resonance-imaging studies (R. Galanello, MD, Cagliari) revealed significant iron overload in the liver; deferoxamine was recommended as the iron-chelating agent least likely to be hepatotoxic.
At the age of 52, after 3 further episodes of severe jaundice, imaging studies showed stenosis of the choledochoduodenostomy with proximal dilatation of the common duct, numerous stones, possible cholangitis, and evidence of iron in the accessory spleen and in the liver. The patient underwent surgery (Dr A.D. Pinna, Bologna) consisting of removing innumerable stones, including a large one, reconstructing the biliary-intestinal anastomosis, and removing the accessory spleen. Immunization against capsulated bacteria was carried out. Since that time the patient has been stable clinically and hematologically, with an Hb of about 11 g/dL, reticulocytes about 4 times the upper normal limit, and mildly elevated bilirubin. The patient, now 58, is head librarian in an academic institution in Rome; he has recently published a book on the liberalism of the Italian philosopher Benedetto Croce. The last time I had contact with him it was to recommend that he receive a COVID-19 vaccination.
Glycolytic enzymopathies
Patient 1 recapitulates the clinical and hematologic features of patients not just with GPI deficiency but with chronic nonspherocytic hemolytic anemia (CNSHA) due to a deficiency of any of the glycolytic enzymes: chronic hemolysis, splenomegaly, gallstones. What varies greatly is the severity of these features, not only from one enzymopathy to another but even from patient to patient within the same enzymopathy. This explains why the age of presentation may range from death in utero to adulthood. The anemia is normocytic and normochromic: if it appears to be macrocytic, it is usually on account of marked reticulocytosis. A shortage of folic acid might also contribute to a high mean corpuscular volume, and since the increased rate of erythropoiesis increases the requirement of this vitamin, folic acid supplementation is recommended. Although there is considerable individual variation in tolerating anemia, in all chronic anemias the body tends to adapt. The best-known red blood cell intrinsic mechanism of adaptation is an increase in bisphosphoglycerate (2,3-DPG) that shifts the Hb-O2 dissociation curve to the right: this can be expected if the flow of glycolysis is impaired downstream, but not upstream, of the Rapoport-Luebering cycle (Figure 1). Indeed, with phosphofructokinase deficiency 2,3-DPG decreases,7 but with pyruvate kinase (PK) deficiency, it increases, and the increased O2 delivery to the tissues is of benefit to the patient.8
The pathophysiology of hemolysis in this group of enzymopathies consists mainly of the removal of red blood cells by macrophages, ie, extravascular hemolysis. That is why splenectomy is so often beneficial (Figure 2). It cannot be claimed to be a radical treatment, but if it raises the Hb level sufficiently to avoid regular blood transfusion, it will also avoid, in most cases, the need for iron chelation therapy, leading to a substantial improvement in quality of life. The accessory spleen was an uncalled-for extra hitch in patient 1: over the years, being a site of hemolysis, it may have become larger than it had been originally, and perhaps that is why it was missed during the first operation.
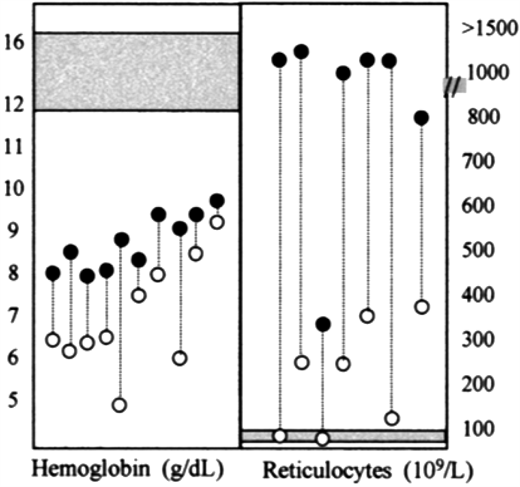
Splenectomy does not cure but does ameliorate CNSHA of PK deficiency. In each of these 10 patients with PK deficiency, a significant increase in the steady-state hemoglobin level was seen after splenectomy (left panel). Whereas in most types of CNSHA an improvement of anemia, resulting from decreased hemolysis, is usually associated with a decrease in the reticulocyte count, in PK deficiency reticulocytes often increase after splenectomy. This paradoxical phenomenon suggests that the spleen selectively removes PK-deficient reticulocytes; the mechanism is not yet well understood.48 From Zanella et al.8
Splenectomy does not cure but does ameliorate CNSHA of PK deficiency. In each of these 10 patients with PK deficiency, a significant increase in the steady-state hemoglobin level was seen after splenectomy (left panel). Whereas in most types of CNSHA an improvement of anemia, resulting from decreased hemolysis, is usually associated with a decrease in the reticulocyte count, in PK deficiency reticulocytes often increase after splenectomy. This paradoxical phenomenon suggests that the spleen selectively removes PK-deficient reticulocytes; the mechanism is not yet well understood.48 From Zanella et al.8
Except for phosphoglycerate kinase deficiency, which is X-linked, all other glycolytic enzymopathies are autosomal recessive: ie, heterozygotes are not affected. This must mean that a 50% reduction in enzyme activity (Figure 3) does not compromise red blood cell survival. Enzymopathy patients are either homozygous for a loss of function mutation, or they have 2 different mutations in the 2 alleles at the same locus (compound heterozygotes, also referred to as biallelic). Whenever a patient is homozygous for a very rare mutation, it is likely that the parents, even if not known to be consanguineous, may be ancestrally related. In other genetic disorders, there are now algorithms aiming to estimate the probability that a particular previously unknown mutation is or is not pathogenic. In the case of enzymopathies, this is not generally necessary because a loss of enzyme activity is in itself a good readout of pathogenicity.

Different phenotypes of heterozygotes for red blood cell enzymopathies. In a heterozygote for PK deficiency, encoded by an autosomal gene (Table 1), the level of enzyme is about one-half of normal in all red blood cells. Since this level of enzyme is sufficient, there are no clinical consequences—ie, PK deficiency is recessive. In a heterozygote for deficiency of G6PD, encoded by an X-linked gene, the situation is quite different: X chromosome inactivation generates red blood cell mosaicism, whereby some red blood cells are entirely normal, and others are G6PD deficient. Therefore, G6PD deficiency is expressed in heterozygotes: it is not recessive.
Different phenotypes of heterozygotes for red blood cell enzymopathies. In a heterozygote for PK deficiency, encoded by an autosomal gene (Table 1), the level of enzyme is about one-half of normal in all red blood cells. Since this level of enzyme is sufficient, there are no clinical consequences—ie, PK deficiency is recessive. In a heterozygote for deficiency of G6PD, encoded by an X-linked gene, the situation is quite different: X chromosome inactivation generates red blood cell mosaicism, whereby some red blood cells are entirely normal, and others are G6PD deficient. Therefore, G6PD deficiency is expressed in heterozygotes: it is not recessive.
The autohemolysis test used in patient 1 was developed by J.V Dacie.9 It was based on the observation that when red blood cells from patients with a wide range of hereditary anemias are incubated in vitro at 37 °C for 24 to 48 hours, some fraction of them undergo hemolysis. If the abnormality is in the membrane, supplying extra glucose usually helps to reduce hemolysis; if the abnormality is instead in glycolysis, glucose does not help. It was a clever test that gave a strong hint—correct in our case—that the patient had an enzymopathy. The test is so economical that in practice it is no longer used. The suspicion of enzymopathy must be raised in any patient with chronic hemolytic anemia presenting early in life, or even later, who does not have a hemoglobinopathy and the morphology characteristic of red blood cell membranopathies; therefore, sound clinical hematology is still paramount. Once the suspicion is formulated, the choice is between a battery of quantitative enzyme assays and a genomic approach. Today the latter is probably preferable: it consists in obtaining from the patient's DNA the sequence of genes whose mutations may cause a red blood cell enzymopathy, or congenital hemolytic anemia in general.10 This approach has been used successfully with a panel of 71 genes,11 at a price (to the Italian National Health Service) of about $950 per patient; several commercial panels are now available. The genomic approach is also advantageous when the patient is heavily transfused, making a red blood cell enzyme assay unreliable.
Among the glycolytic enzymopathies, the one that has been best characterized in terms of management is PK deficiency (red blood cell morphology in Figure 4),12 mainly because it is the most common (although, with an estimated frequency in the United States of 5 per 100 000, it is still a rare disease); therefore, the range of the clinical burden is better known. From the clinical point of view, it is noteworthy that iron overload is frequent, even in patients who are not transfusion dependent.13 This may be due to hepcidin suppression, consistent with a component of ineffective erythropoiesis in the anemia of PK deficiency.14 In this respect it is interesting that, upon diagnostic testing of inherited anemias by sequencing a gene panel, several patients who had been diagnosed with congenital dyserythropoietic anemia had in fact PKLR mutations, ie, PK deficiency.11
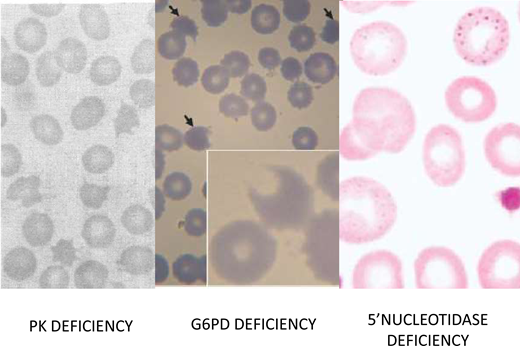
Red blood morphology in select enzymopathies.Left panel: in a patient with CNSHA due to PK deficiency, the blood smear is not diagnostic; however, the presence of “prickle red cells” should raise suspicion. From Mahendra et al.49 Middle panel: “bite cells,” hemighosts and microspherocytes, characteristic of oxidative hemolysis, are seen in this smear from a child who received a dapsone-chlorproguanyl combination for the treatment of acute P. falciparum malaria. From Pamba et al.50 Right panel: red blood cells with fine basophilic stippling in a patient with P5N deficiency. This enzymopathy is the most common inherited cause of basophilic stippling, which is also seen with lead poisoning (lead inhibits many enzymes, including P5N, and it can produce a phenocopy of P5N deficiency). From Rees, Duley, and Marinaki.51
Red blood morphology in select enzymopathies.Left panel: in a patient with CNSHA due to PK deficiency, the blood smear is not diagnostic; however, the presence of “prickle red cells” should raise suspicion. From Mahendra et al.49 Middle panel: “bite cells,” hemighosts and microspherocytes, characteristic of oxidative hemolysis, are seen in this smear from a child who received a dapsone-chlorproguanyl combination for the treatment of acute P. falciparum malaria. From Pamba et al.50 Right panel: red blood cells with fine basophilic stippling in a patient with P5N deficiency. This enzymopathy is the most common inherited cause of basophilic stippling, which is also seen with lead poisoning (lead inhibits many enzymes, including P5N, and it can produce a phenocopy of P5N deficiency). From Rees, Duley, and Marinaki.51
Mitapivat, an allosteric activator of PK (Figure 5), produced a significant increase in hemoglobin level in one-half of 52 patients with PK deficiency in a phase 3 multicentric clinical trial; this was associated with a decrease in bilirubin and in reticulocytes.15 Mitapivat may be on track to become the first approved drug for a red blood cell enzymopathy.
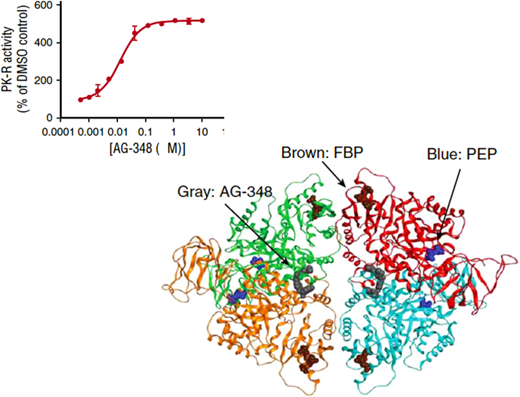
Mitapivat is an allosteric activator of PK.Top panel: the sigmoid-shaped dependence of PK activity on the concentration of AG-348 (mitapivat) suggests cooperative interaction among the PK subunits. Bottom panel: diagram of the three-dimensional structure of the PK tetramer with bound mitapivat (gray). Blue: the substrate phosphoenolpyruvate PEP; brown: the physiological activator fructose-1,6-bisphosphate (FBP). Modified from Kung et al.52
Mitapivat is an allosteric activator of PK.Top panel: the sigmoid-shaped dependence of PK activity on the concentration of AG-348 (mitapivat) suggests cooperative interaction among the PK subunits. Bottom panel: diagram of the three-dimensional structure of the PK tetramer with bound mitapivat (gray). Blue: the substrate phosphoenolpyruvate PEP; brown: the physiological activator fructose-1,6-bisphosphate (FBP). Modified from Kung et al.52
In the management of patient 1, at least 7 senior colleagues from 4 different countries with specialized expertise in different areas have been involved: I found their help invaluable. At the same time, it must be noted that a person with a potentially disabling disease can still hold on and have a quality of life that enables him to contribute to the life and culture of others.
CLINICAL CASE
A 3-year-old girl from Egypt was seen in London for an assessment of her clinical state. Her parents also requested genetic counseling. The patient was the couple's firstborn. Her height and weight were in the lower 10th percentile, and she was thin but did not appear malnourished. She had bilateral choreoathetosis, involuntary muscle movements and spasms, and upward eye movements; her speech was limited and dysarthric. Her history was highly significant in that she had been born at 35 weeks' gestation and at birth had had severe jaundice, but no hospital records were available. On one occasion after eating fava beans, she had become lethargic and had passed dark urine. At the time of her visit, her blood count, including reticulocytes, was normal: there was no evidence of CNSHA. The 3 members of the family were all Rh+, and there was no ABO incompatibility setup. Red blood cell glucose-6-phosphate dehydrogenase (G6PD) assays yielded values of 1.5 in the patient, 7.2 in her mother, and 0.6 in her father (normal values, 7-10 IU/g Hb). The most likely diagnosis was severe neurological damage from kernicterus. We inferred from the G6PD results that the father was hemizygous G6PD deficient: sequence analysis of his G6PD gene identified a previously unknown mutation, p.N135T, that was hence named G6PD Cairo; the daughter was heterozygous and the mother, normal. The mode of inheritance of G6PD deficiency and its implications were explained to the family. Father and daughter were advised not to eat fava beans. We informed them that the problem of neonatal jaundice (NNJ) was unlikely to recur if a subsequent baby were male, but unfortunately, it might recur in a female, although it might be less severe. We emphasized the importance of having the baby in a location where NNJ could be appropriately managed.
Redox enzymopathies
The most prominent enzymopathy in this group is indeed G6PD deficiency.16 From an epidemiological point of view, whereas the glycolytic enzymopathies are all rare to ultrarare, here we are at the other end of the spectrum: G6PD deficiency is present in an estimated 500 million people worldwide (Figure 6). From a clinical point of view, we are dealing with a very different situation. Whereas the glycolytic enzymopathies cause lifelong CNSHA, G6PD deficiency in itself does not cause anemia and is not even a disease; rather, it is a genetic abnormality that in most cases remains asymptomatic for a lifetime (except for the very rare variants that cause CNSHA, clinically similar to that from glycolytic enzymopathies16 ). However, it can manifest at birth through NNJ (as in patient 2) or at any age,17 like a thunderbolt out of a blue sky, in the form of acute hemolytic anemia (AHA), when the person is challenged by an agent that causes oxidative damage to the red blood cells (Figure 4).18 The agent may be the ingestion of fava beans, or infection, or a drug (probably in that order of frequency: for the mechanism, see Figure 7).19 In G6PD-deficient persons, AHA is a prototype example of a gene-environment interaction causing a disease that can be life-threatening. Favism in children can be fatal if not promptly treated, and rasburicase, potentially a lifesaver in a patient with an incumbent tumor lysis syndrome, can become a serious hazard if the patient happens to be G6PD deficient (Figure 7B)—a typical example of a pharmacogenetic hazard.20

Epidemiology of G6PD deficiency throughout the world. Each country on the map is shaded in a color based on the best estimate of the mean frequency of G6PD deficiency allele(s) in that country (this is the same as the frequency of G6PD-deficient males). The larger panel gives a color-coded list of 10 common G6PD variants associated with G6PD deficiency: asterisk-shaped symbols in the corresponding colors are shown in the countries where these variants have been observed (for graphic reasons symbols could not be inserted in all countries where the respective variants are present). Modified from Luzzatto, Ally, and Notaro.16
Epidemiology of G6PD deficiency throughout the world. Each country on the map is shaded in a color based on the best estimate of the mean frequency of G6PD deficiency allele(s) in that country (this is the same as the frequency of G6PD-deficient males). The larger panel gives a color-coded list of 10 common G6PD variants associated with G6PD deficiency: asterisk-shaped symbols in the corresponding colors are shown in the countries where these variants have been observed (for graphic reasons symbols could not be inserted in all countries where the respective variants are present). Modified from Luzzatto, Ally, and Notaro.16
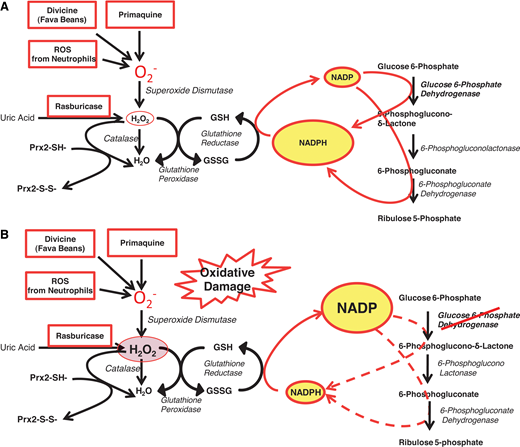
The role of G6PD in protecting red blood cells from oxidative damage. (A) In G6PD-normal red blood cells, G6PD and 6-phosphogluconate dehydrogenase—2 of the enzymes of the PPP—provide an ample supply of NADPH, which in turn regenerates GSH when this is oxidized by ROS (eg, superoxide, O2− and H2O2). Thus, when O2− (meant here to represent itself and other ROS) is produced by pro-oxidant compounds such as primaquine, or the glucosides in fava beans (divicine), or the oxidative burst of neutrophils, these ROS are rapidly neutralized; similarly, when rasburicase administered to degrade uric acid produces an equimolar amount of H2O2, this is rapidly degraded by the combined action of GSH peroxidase, catalase, and Prx2 (peroxiredoxin-2: all 3 mechanisms are NADPH dependent). (B) In G6PD-deficient red blood cells, where the enzyme activity is reduced, NADPH production is limited, and it may not be sufficient to cope with the excess ROS generated by pro-oxidant compounds and the consequent excess H2O2. This diagram also explains why a defect in GSH reductase has very similar consequences to G6PD deficiency. Modified from Luzzatto, Nannelli, and Notaro.53 PPP, pentose phosphate pathway.
The role of G6PD in protecting red blood cells from oxidative damage. (A) In G6PD-normal red blood cells, G6PD and 6-phosphogluconate dehydrogenase—2 of the enzymes of the PPP—provide an ample supply of NADPH, which in turn regenerates GSH when this is oxidized by ROS (eg, superoxide, O2− and H2O2). Thus, when O2− (meant here to represent itself and other ROS) is produced by pro-oxidant compounds such as primaquine, or the glucosides in fava beans (divicine), or the oxidative burst of neutrophils, these ROS are rapidly neutralized; similarly, when rasburicase administered to degrade uric acid produces an equimolar amount of H2O2, this is rapidly degraded by the combined action of GSH peroxidase, catalase, and Prx2 (peroxiredoxin-2: all 3 mechanisms are NADPH dependent). (B) In G6PD-deficient red blood cells, where the enzyme activity is reduced, NADPH production is limited, and it may not be sufficient to cope with the excess ROS generated by pro-oxidant compounds and the consequent excess H2O2. This diagram also explains why a defect in GSH reductase has very similar consequences to G6PD deficiency. Modified from Luzzatto, Nannelli, and Notaro.53 PPP, pentose phosphate pathway.
Patient 2 illustrates that NNJ, a clinical manifestation of G6PD deficiency known for decades,21,22 is no less important than AHA. In most G6PD deficient babies, the hyperbilirubinemia is mild: it overlaps with “physiological jaundice” of the newborn. However, in some cases, like this one, it was severe enough to cause kernicterus. In many countries, G6PD deficiency is the most common cause of severe NNJ: in 1 study in Nigeria among babies admitted to the hospital after showing clinical signs of kernicterus, the frequency of G6PD deficiency was 78%—compared to 22% in the general population.23 The underlying G6PD variant was G6PD A−, the same that accounts for many cases of NNJ, including severe ones, in the United States and one of the reasons why neonatal screening for G6PD deficiency has been advocated.17,24 The reasons for severity may be environmental (eg, infection, exposure to naphthalene) or genetic (coexistence of 1 or 2 UGT1A1 mutant alleles),25 and they are in part unknown. The management of G6PD-related NNJ is no different from that due to other causes: this child should have had exchange blood transfusion, but unfortunately, she did not. G6PD Cairo, the G6PD variant first identified in patient 2, was subsequently found to be quite common in Egypt and elsewhere, particularly in patients presenting with acute favism.26
The importance of G6PD being X-linked cannot be overemphasized. First, since males have only 1 X chromosome, they are either hemizygous G6PD normal or hemizygous G6PD deficient; females can be homozygous normal or G6PD deficient (biallelic, whether homozygous or compound heterozygous) or heterozygous for G6PD deficiency (ie, with 1 normal allele). Second, in any population, according to the Hardy-Weinberg rule, heterozygous females are more common than hemizygous G6PD-deficient males (Table 2). Third, since G6PD is subject to X chromosome inactivation, in heterozygous females there is red blood cell mosaicism, whereby a proportion of red blood cells are just as deficient in G6PD activity as in a hemizygous male (Figure 3). In heterozygotes the expression of G6PD deficiency is highly variable, and since X inactivation can be randomly “skewed,” their enzyme levels range from normal to markedly deficient so that in an individual female the proportion of G6PD deficient red blood cells may be high (even up to 90%). This last point is clinically relevant, as it was unfortunately in our patient 2.
G6PD deficiency in several sample populations
| Examples | Frequency of G6PD deficiency in (hemizygous) males, % | Frequency of G6PD deficiency in females, % | |
|---|---|---|---|
| Heterozygous | Homozygous | ||
| US | 2.5 | 4.9 | 0.06 |
| Nigeria | 22 | 34 | 4.8 |
| Sardinia | 12 | 21 | 1.4 |
| Thailand | 7.3 | 13.5 | 0.5 |
| Examples | Frequency of G6PD deficiency in (hemizygous) males, % | Frequency of G6PD deficiency in females, % | |
|---|---|---|---|
| Heterozygous | Homozygous | ||
| US | 2.5 | 4.9 | 0.06 |
| Nigeria | 22 | 34 | 4.8 |
| Sardinia | 12 | 21 | 1.4 |
| Thailand | 7.3 | 13.5 | 0.5 |
Frequency in US from Chinevere et al44 ; in Nigeria, from Luzzatto and Allan45 ; in Sardinia, from Cocco et al46 ; in Thailand, from Bancone et al.47 The frequency of G6PD deficiency in hemizygous males is identical to the frequency of the G6PD mutant allele (or the sum total of the frequencies if more than 1 mutant allele is involved). From the male frequency, it is easy to calculate the frequencies of the female genotypes, on the assumption that the population is in Hardy-Weinberg equilibrium. The data in this table show that, as all textbooks say, homozygous females are few compared to hemizygous males; on the other hand, heterozygous females are almost twice as common as hemizygous males (which most textbooks fail to say). Because of epigenetic mosaicism consequent on X chromosome inactivation, all heterozygotes have some G6PD deficient red blood cells: on average 50%, but with a wide range of variation. This is why in heterozygotes clinical manifestations are, on average, less severe, but not always so. In every series of patients with favism, there are always heterozygous girls.19
Given the high frequency of G6PD deficiency in many parts of the world (Figure 6), blood donors may be G6PD deficient. A recent careful investigation has found that after blood bank storage for 6 weeks the majority of G6PD-deficient blood units still meet, 24 hours after transfusion, the in vivo red blood cell survival target of >75%.27
Some ultrarare enzymopathies in the redox category involve either the biosynthesis or the function of GSH (Table 1). GSH reductase deficiency can manifest as favism,28 confirming the key role of GSH in the defense of red blood cells against oxidative stress.
Cytochrome b5 reductase (CBR) deficiency is an enzymopathy that does not cause hemolytic anemia: it instead causes recessive congenital methemoglobinemia,29 often presenting as cyanosis in infants, that may mislead one to suspect congenital heart disease. This NADH-dependent enzyme (formerly known as diaphorase), capable of reducing methemoglobin (Fe+++) back to hemoglobin (Fe++), is encoded by CYB5R3.* Most missense mutations in this gene cause only methemoglobinemia (type I disease), whereas nonsense mutations, deletions, and some specific missense mutations cause, in addition, a devastating neurological syndrome (type 2 disease, probably mediated by the role of the enzyme in the biosynthesis of myelin phospholipids). A point of practical importance is that in patients with methemoglobinemia (whether from CRB deficiency or from hemoglobin M) pulse oximetry measured with bedside devices is not reliable.
CLINICAL CASE
A 12-year-old boy from Izmir, Turkey, the firstborn from first-cousin parents with normal blood counts, was referred to an academic hematology department because of hemolytic anemia diagnosed at the age of 2. The anemia was moderate most of the time but with exacerbations, concomitant to infection, that sometimes required blood transfusion. Physical examination revealed pallor, jaundice, hepatomegaly (2 cm below the costal border) and splenomegaly (4cm below the costal border). He had anemia, with a hemoglobin of 7.5 g/dL and a reticulocytosis of 160 × 109/L. White blood cell count, platelets, and liver enzymes were within the normal range. A direct Coombs test was negative. The patient showed an increased unconjugated bilirubin of 2.1 mg/dL (upper normal limit 0.8 mg/dL) and lactate dehydrogenase 678 IU (upper normal limit 300). Haptoglobin was undetectable. A peripheral blood smear revealed anisopoikilocytosis, macrocytes, polychromatic cells, rare elliptocytes and spherocytes, and basophilic stippling (Figure 4). Hemoglobin electrophoresis was normal, and a test for unstable hemoglobin was negative. Folic acid and vitamin B12 levels were normal. An osmotic fragility test was normal. A bone marrow aspirate showed erythroid hyperplasia; an iron stain did not reveal sideroblasts. An enzyme assay panel yielded normal values of glycolytic enzymes and of G6PD. The ultraviolet spectrum of an extract of red blood cell metabolites showed an abnormal purine/pyrimidine ratio, suggestive of pyrimidine 5′-nucleotidase (P5N) deficiency. Sequencing of the NT5C3 gene in the patient's DNA revealed homozygosity for a frameshift mutation (c.393-394del TA, K132Rfs7*). Both parents were heterozygous for the same mutation.31
Courtesy of Dr Paola Bianchi.
Nucleotide metabolism enzymopathies
P5N deficiency, although rare, ranks third in frequency among enzymopathies (after G6PD deficiency and PK deficiency).32 P5N, encoded by the gene NT5C3A, is particularly important during the last stage of erythroid maturation. Reticulocytes, having no RNA synthesis, are unable to recycle pyrimidine nucleotides arising from RNA degradation (as would be the case in other cells), and these must be hydrolyzed to nucleosides before they can exit through the membrane. With P5N deficient, encumbrance by pyrimidine nucleotides seems to hinder the ability of reticulocytes to get rid of their last ribosomes or of their remnants. These become visible under the shape of basophilic stippling,33 a morphological abnormality characteristic of P5N deficiency (Figure 4) but not unique to it (see patient 1). The hematologic consequences of this enzymopathy are illustrated by patient 3, who has a moderately severe CNSHA.
The other (ultrarare) enzymopathy in this group is adenylate kinase (AK) deficiency. AK, by catalyzing the interconversion of ADP into ATP and adenosine mono-phosphate (AMP), is generally important in controlling the level of these compounds.3 In red blood cells ATP is crucial for the cation pump, and AMP, a precursor of the signaling molecule cyclic AMP,3 may be important in maintaining erythrocyte deformability.34 Indeed, in AK deficiency there is evidence of intravascular hemolysis.35 For both P5N and AK, it is through their respective genetic defects that we know they have an essential function in red blood cells.
Conclusion
Red blood cell enzymopathies are now understood at the biochemical and molecular levels, and their clinical and hematologic features are reasonably well characterized. G6PD deficiency stands out in terms of epidemiology because it is a widespread, mostly asymptomatic abnormality: it bears witness to the role of malaria in shaping human evolution, and its clinical manifestations are largely preventable and manageable. Although recognizing chronic hemolytic anemia is not difficult, the other enzymopathies are probably still underdiagnosed; DNA testing by gene panels should gradually overcome this diagnostic gap. In contrast, the targeted treatment of red blood cell enzymopathies is nearly uncharted territory. Since most of them qualify as orphan diseases, there are incentives to developing new therapies.
Acknowledgments
I have been fortunate to work on red blood cell enzymopathies, particularly G6PD deficiency, in 5 countries. Since I cannot list all those to whom I am grateful, I will only mention Olugbemiro Sodeinde (Ibadan), the late Graziella Persico (Naples), Tom Vulliamy (London), Letizia Longo (New York), Rosario Notaro (Florence), and Stella Malangahe (Dar es Salaam). Research support was received from the World Health Organization, National Research Council (Italy), Medical Research Council (UK), and National Institutes of Health (in Nigeria and in the US). I am especially grateful to all patients with enzymopathies for what I have learned from them.
Conflict-of-interest disclosure
Lucio Luzzatto: no competing financial interests to declare.
Off-label drug use
Lucio Luzzatto: nothing to disclose.
In red blood cells there is also an NADPH-dependent enzyme that can reduce methemoglobin back to hemoglobin: it was formerly called flavin reductase and is now known to be identical to biliverdin-IX reductase.30 Once again it is from genetic evidence that we know that CBR is the enzyme responsible for the physiologic maintenance of hemoglobin in the Fe++ state since mutations of CYB5R3 cause methemoglobinemia, whereas mutations of BLVRB do not.
