

Abstract
The landscape of sickle cell disease (SCD) treatment continues to evolve rapidly, with new disease-modifying therapies in development and potentially curative options on the horizon. Until recently, allogeneic stem cell transplant has been the only proven cure for SCD. Gene therapy is rising to the forefront of the discussion as a potentially curative or highly disease- modifying option for abating the complications of the disease. Understanding the different types of gene therapy in use, the differences in their end points, and their potential risks and benefits will be key to optimizing the long-term use of this therapy.
Learning Objectives
Have an improved understanding of the different types of gene therapy
Be able to have a more thoughtful conversation with patients regarding gene therapy
CLINICAL CASE
A 22-year-old woman with sickle cell anemia (HbSS disease) presents to you, a sickle cell specialist (as a referral from her community hematologist-oncologist), to discuss the possibilities of curative treatment. She has a history of frequent vaso-occlusive (VOC) crises and goes to the emergency department about 5 times per year, where she is admitted most of those times. She has never had a stroke (that she is aware of). She has been on hydroxyurea (HU), 1000 mg per day, since she was 9, pretty reliably. She takes 30 mg of morphine sulfate extended release tablets (MS Contin, Purdue) taken by mouth twice a day, 10 mg of oxycodone every 4 hours as needed, and folic acid (in addition to HU). She is on no other medications. She has no full siblings and does not know her father. She lives with her mother and 2 half siblings approximately 2 hours from your clinic.
On exam, she weighs 60 kg, with a normal body mass index, a heart rate of 75, a respiration rate of 20, a blood oxygen level at 93%, and a blood pressure reading of 118/70.
Sickle cell disease (SCD) has been well characterized for over 100 years, with the first clinical report published in 1910 describing it as the “first molecular disease.”1 Despite this long scientific history, progress toward identifying a cure has been slow, likely due in part to the fact that SCD affects mostly individuals living in low-resource settings or minority populations living in high-resource nations. Further, the development of disease-modifying therapies has also been sluggish, with only 1 medication available until 2017.
SCD results from the inheritance of at least 1 copy of sickle hemoglobin (HbS) and a second copy of a gene encoding HbS or another abnormal hemoglobin. HbS is produced when the β-globin gene (HBB) contains a single E6V missense mutation resulting in the replacement of β6 glutamic acid by valine.2 On deoxygenation, HbS polymerizes, leading to abnormally shaped red cells and multiple downstream clinical sequelae, including hemolysis, vaso-occlusion and subsequent progressive and irreversible organ damage, decreased quality of life, and early death.3 Although HbS polymerization, vaso-occlusion, and hemolytic anemia are central to the pathophysiology of SCD, the resultant pathological events are more consistent with secondary vascular-endothelial dysfunction and widespread inflammation such that the complications of SCD are better understood as inflammatory vascular disorders.
The source of SCD pathology remains a single-point mutation resulting in the production of abnormal protein (HbS) and subsequent hemoglobin polymerization within the red blood cell. The rate of HbS polymerization is highly variable and depends on multiple factors, including the amount of HbS per erythrocyte, the amount of other nonsickling hemoglobin per erythrocyte, and the overall erythrocyte hemoglobin level. Thus, sufficient healthy, normal adult hemoglobin (HbA) in each cell, as in individuals who carry the sickle cell trait (HbAS), will prevent polymerization and reduce the symptoms and sequelae of the disease except under rare instances of extreme physiological stress.4,5 Similarly, fetal hemoglobin (HbF) has an antisickling effect and can reduce or possibly eliminate hemoglobin polymerization if in sufficient quantity within the erythrocyte.6
Four different SCD-specific medications have now been approved to treat the effects of SCD. HU, the first approved, has been proven to decrease the frequency of VOC, reduce stroke risk in some affected patients, and improve the quality and length of life in many affected individuals.7 However, HU has not been proven as a universally acceptable disease-modifying therapy because many individuals continue to develop significant disease- related complications or are unwilling to maintain drug adherence due to concerns with side effects, fertility, or tolerance. L-glutamine, approved in 2017 to treat SCD, has antioxidant properties that have been shown to ameliorate SCD and decrease the frequency of VOC.8 Long-term durability and utility have yet to be demonstrated. The newest medications, crizanlizumab and voxelotor, have proven efficacious in their own right by decreasing VOCs and decreasing hemolysis, respectively, but neither medication is likely to have a demonstrable curative effect, and both require ongoing, lifelong treatment.9,10 Further, these medications are not yet proven to reduce, delay, or stabilize organ-specific complications such as renal disease, avascular necrosis, or pulmonary hypertension.
CLINICAL CASE (continued)
You review her current disease status, complications, and concerns and specifically discuss the importance of doing a full screening assessment of all organ systems to evaluate the SCD- related complications. You perform a full set of labs, an evaluation for iron overload (ferritin, liver magnetic resonance imaging), an echocardiogram (due to low oxygen at baseline), a workup for alloimmunization, and a kidney screen (albumin/creatinine ratio). Her lab results are consistent with HU adherence, and the echocardiogram shows trivial tricuspid regurgitation and mild left ventricular hypertrophy but is otherwise normal. Labs show that ferritin is 1200 µg/L, liver iron concentration is 5.1 mg/g, and there is no concern for significant alloimmunization. Unfortunately, her renal assessment shows marked proteinuria.
You discuss treatment options with the patient, including potentially increasing HU to the maximum tolerated dose or adding additional medical therapies. In frustration, however, she says, “I want to be cured of this wretched disease!”
One method by which to correct the pathophysiological abnormality in SCD is to replace the abnormal HbS with more functional HbA. The clinical benefit of this molecular replacement has been clearly demonstrated by the success of hematopoietic stem cell transplant (HSCT) for SCD. In this scenario, new hematopoietic stem cells (HSCs) are used as the vehicle to deliver healthy hemoglobin and therefore eliminate erythrocyte sickling and the secondary effects of this pathological process.11,12 Multiple studies have demonstrated the efficacy of HSCT for SCD, including donor HSC with HbAS.13
While highly efficacious, HSCT is not an option for all individuals living with SCD. The best outcomes reported are in younger individuals with SCD who have a matched sibling donor; unfortunately, relatively few individuals with SCD have such a donor.14 Improvements in haploidentical HSCT for SCD are rapidly evolving and will significantly increase the available donor pool. However, there are still potential drawbacks that can include graft failure, delayed immune reconstitution, infertility, secondary malignancy, and graft-versus-host disease (GVHD).15 Further, it is unlikely any allogeneic transplant could completely remove the risk of GVHD disease or the need for long-term immune suppression therapy, both of which carry their own risks of subsequent complications. Thus, there remains a demonstrated need for other means of providing gene transfer into HSCs without the same immunologic risks. In this scenario, transplantation of genetically modified autologous HSCs provides a potential alternative therapy.
CLINICAL CASE (continued)
You discuss potentially curative options with your patient that include haploidentical HSCT and gene therapy, as you have already determined that she does not have any full siblings. Her mother works full time and has a history of lupus that is on active treatment (and so cannot donate HSCs). She is interested in considering haploidentical transplant from a sibling but is worried about the distance from home (weekly appointments to modulate her immune suppression, the need to stay in close proximity after transplant, a concern for GVHD and subsequent rehospitalization). Using shared decision-making, it seems she may be best suited for gene therapy, as there is no risk of GVHD and no need to monitor immune suppression medication. She asks about the different types of gene therapies available.
Broadly speaking, 4 main types of gene therapy are available for the treatment of SCD. These include gene addition therapy, gene editing, gene silencing, and gene correction therapy (Figure 1). Each type of therapy differs in the means by which it induces the replacement of HbS with nonsickling hemoglobin. The nonsickling hemoglobin is the target protein in each therapy and will need to be evaluated using multiple novel methods and terms.
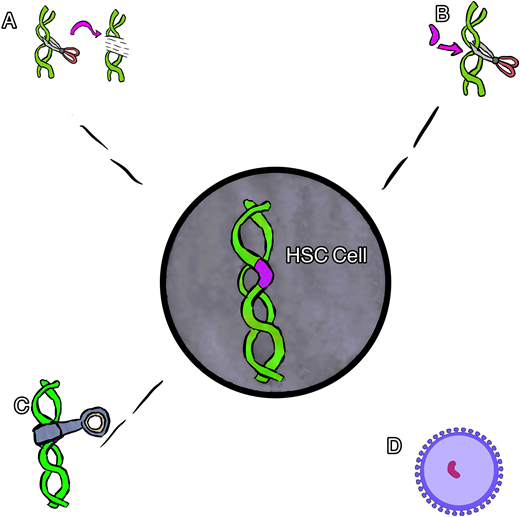
Cartoon rendering of different gene therapies. (A) Gene editing, (B) gene correction, (C) gene silencing, and (D) gene addition via viral vector.
Cartoon rendering of different gene therapies. (A) Gene editing, (B) gene correction, (C) gene silencing, and (D) gene addition via viral vector.
Gene addition therapy is the addition of a new gene using a viral vector (usually) to deliver a nonsickling globin gene to the stem cells. In this procedure the native HbS gene is not altered, resulting in the production of both the new hemoglobin and the native HbS. Several examples of gene addition therapy are ongoing that use a lentiviral vector (LVV) to house and deliver a new gene.16
Gene editing is most often used to describe a process of gene disruption in the context of SCD and can be used to target suppressors of HbF as a way to both increase HbF and decrease HbS. Elements of DNA within a gene can be targeted using a guide that can identify and tightly bind to the target with high specificity coupled with an enzyme to cut the DNA, inducing double-stranded breaks. The specific DNA cut allows one to change the sequence with high precision, usually resulting in an insertion and deletion. This type of gene therapy most often targets a different part of DNA (separate from the HbS mutation) to produce an increase in HbF production while reciprocally suppressing HbS production.16 Specifically, many of the current therapies target the BCL11A gene, a negative regulator of HbF. In this example, the gene editing is used to turn off the regulation of HbF in order to increase HbF production.
Gene silencing uses the regulation of gene expression in a cell to prevent the expression and resultant production of certain proteins. Similar to gene editing, this type of therapy is being used to suppress the BC11A gene, resulting in an increase in HbF while reciprocally suppressing HbS production. In contrast to gene editing, this type of gene therapy has thus far relied upon viral vector delivery (similar to gene addition) to deliver an antisense to messenger RNA to suppress the gene product instead of cutting the gene.17
Gene correction can be performed in several different ways. In most cases, however, a guide RNA is used to identify the target mutation for cutting, and then editing occurs with the simultaneous delivery of template DNA of the correct sequence, directing homology-directed repair (HDR; Figure 2). Though this is currently the least efficient method, efforts are underway to improve gene correction, including through the insertion of DNA, direct base editing, and prime editing. This is the only type of gene therapy that currently aims to eliminate HbS production and introduce a nonsickling hemoglobin simultaneously.18
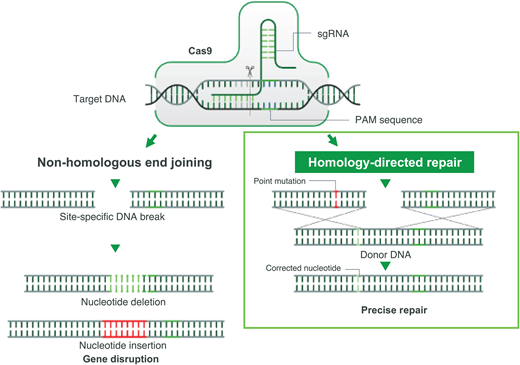
CRISPR-Cas-9-induced double-stranded break and its sequential repair pathways.Left: nonhomologous end joining. Right: HDR, which requires the insertion of a homologous DNA strand used as a template for a high-fidelity double-stranded DNA break. PAM, protospacer adjacent motif; sgRNA, single-guide RNA.
CRISPR-Cas-9-induced double-stranded break and its sequential repair pathways.Left: nonhomologous end joining. Right: HDR, which requires the insertion of a homologous DNA strand used as a template for a high-fidelity double-stranded DNA break. PAM, protospacer adjacent motif; sgRNA, single-guide RNA.
At present, all types of gene therapy use the same overall procedure. For each study listed, patients first undergo intensive screening. It is important to note that the criteria for each study differ slightly, but all require the individual to have had significant SCD-related complications (as a reason for undergoing a study procedure) and to have sufficient organ function to undergo the chemotherapy preparation required. Once fully screened, patients need to undergo stem cell collection using plerixafor mobilization and apheresis. This process may be undertaken more than once to ensure sufficient stem cells are collected for manipulation as well as for backup. Once the stem cells are appropriately altered, all types of gene therapy utilize chemotherapy (to make room for altered/manipulated stem cells). In all but 1 trial below (MOMENTUM), these studies use busulfan chemotherapy to provide myeloablation to ensure optimal stem cell engraftment. The MOMENTUM trial uses a “reduced-intensity” chemotherapy regimen using melphalan.
Current and upcoming clinical trials using gene therapy are detailed in Table 1. Note that some of the trials listed target the BCL11A gene (its erythroid enhancer of messenger RNA), a repressor of γ-globin expression, in order to induce HbF production in adult erythrocytes, while others use random viral vector insertion to result in HbA production, and 1 study, notably, targets the sickle cell gene mutation itself.17-20
Current and upcoming studies of gene therapy in SCD
| Study name | LentiGlobin | DREPAGLOBE | CLIMB | PRECIZN-1 | Genetic silencing of BCL11A | MOMENTUM | CEDAR |
|---|---|---|---|---|---|---|---|
| Type of gene therapy | Gene addition | Gene addition | Gene editing | Gene editing | Gene silencing | Gene addition | Gene correction |
| Editing tool | NA | NA | CRISPR-Cas9 RNP | Zinc finger | ShRNA | NA | HiFi CRISPR-Cas9 RNP |
| Type of stem cell manipulation | Transduction | Transduction | Electroporation | Transfection with zinc finger nuclease mRNA | Transduction | Transduction | Electroporation |
| Vector (y/n) | BB305 LVV | DROBE 1 LVV | None | None | BCH-BB694 LVV that encodes a microRNA-adapted shRNA | γG16D LVV | Nonintegrating AAV6 donor DNA repair template |
| Genetic target (y/n) | NA | NA | Erythroid lineage- specific enhancer of the BCL11A gene | 11A (BCL11A) locus (erythroid enhancer) | BCL11A mRNA | N/a | Sickle mutation (adenosine— > thymine [A— > T] |
| Drug product | LentiGlobin BB305 | DREPAGLOBE | CTX001 | BIVV003 | BCH-BB694 | ARU-180126 | GPH101 |
| Protein product | HbAT87Q | βAS3, an antisickling β-globin protein (AS3) containing 3 amino acid substitutions in the wild-type HBB | HbF | HbF | HbF | HbFG16D | HbA |
| Study name | LentiGlobin | DREPAGLOBE | CLIMB | PRECIZN-1 | Genetic silencing of BCL11A | MOMENTUM | CEDAR |
|---|---|---|---|---|---|---|---|
| Type of gene therapy | Gene addition | Gene addition | Gene editing | Gene editing | Gene silencing | Gene addition | Gene correction |
| Editing tool | NA | NA | CRISPR-Cas9 RNP | Zinc finger | ShRNA | NA | HiFi CRISPR-Cas9 RNP |
| Type of stem cell manipulation | Transduction | Transduction | Electroporation | Transfection with zinc finger nuclease mRNA | Transduction | Transduction | Electroporation |
| Vector (y/n) | BB305 LVV | DROBE 1 LVV | None | None | BCH-BB694 LVV that encodes a microRNA-adapted shRNA | γG16D LVV | Nonintegrating AAV6 donor DNA repair template |
| Genetic target (y/n) | NA | NA | Erythroid lineage- specific enhancer of the BCL11A gene | 11A (BCL11A) locus (erythroid enhancer) | BCL11A mRNA | N/a | Sickle mutation (adenosine— > thymine [A— > T] |
| Drug product | LentiGlobin BB305 | DREPAGLOBE | CTX001 | BIVV003 | BCH-BB694 | ARU-180126 | GPH101 |
| Protein product | HbAT87Q | βAS3, an antisickling β-globin protein (AS3) containing 3 amino acid substitutions in the wild-type HBB | HbF | HbF | HbF | HbFG16D | HbA |
Current clinical trials of lentiviral gene therapy based on the addition of a modified β-globin gene (HbAT87Q) have accumulated the most data so far and have demonstrated a benefit in the reduction of significant VOCs in SCD. However, the data remain early, and results regarding improvements in long-term durability and organ function are forthcoming. CRISPR Therapeutics and Vertex Pharmaceuticals and investigators from Boston Children's Hospital have also presented data on their CLIMB and short hairpin RNA (shRNA) studies, respectively, showing that the potent antisickling properties of HbF combined with the lower levels of HbS result in the resolution of VOCs as well. These gene editing and silencing studies, however, are even earlier in their reporting and long-term outcomes.
Gene correction therapy will be the next type of gene therapy to enter the clinical space. Gene correction therapy includes a combination of gene editing and gene addition. The CEDAR study, which received US Food and Drug Administration clearance to move forward to a phase 1 clinical trial in early 2021, will concurrently use a high-fidelity clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 ribonucleoprotein (RNP) to induce a double-strand DNA breakage and HDR with a nonintegrating adeno-associated virus-6 donor DNA repair template to produce a new gene product. Unlike the gene-editing therapies that rely on nonhomologous end joining and subsequent insertion and deletion formation in the edited space, gene correction therapies rely on the more complicated and historically less efficient HDR.21
CLINICAL CASE (continued)
You and your resident leave the room to allow the patient and her family time to discuss and digest all of this information. Your resident astutely asks how it will be possible to compare outcomes and measure efficacy in trials that use different methods of hemoglobin induction and different types of hemoglobin production.
There are several ways to evaluate efficacy in gene therapy for SCD. At the end of the process, the most important question is how much of the new hemoglobin (protein product of the gene therapy) is being produced over time. However, throughout the process are many steps in which interim evaluations of efficacy are useful. These steps and their evaluation techniques are detailed in Figure 3.
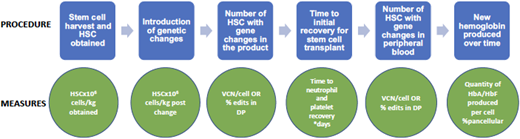
Gene therapy process and evaluation measures. DP, drug product; VCN, vector copy number.
Gene therapy process and evaluation measures. DP, drug product; VCN, vector copy number.
As noted, it is most important to determine how much nonsickling hemoglobin is in each red blood cell and how much of it is a product of the gene therapy vs the myeloablation (which can result in stress erythropoiesis that causes HbF production). This can be evaluated by the transduction efficiency, or the percentage of blood stem cells having incorporated the desired genetic material. Longitudinal studies are needed to determine the durability of gene therapy. Last, it will be important to learn over time which symptoms and/or complications of SCD improve with gene therapy and if the outcomes differ depending on the type and amount of hemoglobin product. At this time it is not clear what percentage of stem cells must receive genetic corrections to result in sufficient nonsickling hemoglobin. Additional follow-up assessments need to include both laboratory and rheologic measures of hemolysis and adhesion in addition to in-depth patient-reported outcomes.
Finally, it is most important to assess whether gene therapy can prevent vaso-occlusion (which types and to what extent) and can either stabilize or resolve organ complications due to SCD. These findings remain unclear at this time but are highly necessary in advancing outcomes in SCD. For example, up to 10% of persons with sickle cell anemia may develop end-stage renal disease. At this time it is unclear whether even allogeneic transplant can prevent the development of end-stage renal disease once someone has developed chronic kidney disease; gene therapy results are further behind. While data regarding outcomes for VOC appear clearer, the long-term organ-specific response to gene therapy will truly measure its efficacy.
CLINICAL CASE (continued)
You and the resident return to the patient and her family. She does not have children but does want to have a family later in life. She is worried about the potential for infertility. Further, she has read about a patient who got leukemia after early-stage gene therapy and wants to know the risks of gene therapy.
There are real and potential risks involved with gene therapy of all types. The chemotherapy used in myeloablation carries a high risk of infertility (nearly 100%) and also results in mucositis, nausea, loss of appetite, alopecia, and other usually reversible complications. Infertility is a major source of concern that must be addressed. While fertility preservation is possible for some individuals, it is neither universally available nor efficacious. It is important to have patients meet with fertility specialists to review the available options and their associated risks and benefits. Secondary malignancy is another major risk of gene therapy. Chemotherapy such as busulfan carries an independent long-term risk of secondary malignancy in patients undergoing both allogeneic and autologous transplant. Another potential cause of a secondary malignancy is the transplantation of potentially damaged HSCs. Individuals with SCD are affected by chronic inflammation and endothelial damage as well as hypoxic bone infarction and constant erythropoietic stress.22 These manifestations of SCD likely damage the HSCs and may result in a predisposition to malignant transformation. At this point it is unclear how high this risk is or if it can be suitably mitigated with changes already in use in gene therapy protocols or future changes to come. Two patients in the initial LentiGlobin HGB-206 trial developed acute myelogenous leukemia at 3 and 5 years post autologous gene therapy.23 Currently, the workup suggests that the vector is not associated with the malignancy and that perhaps there is an inherent increased risk in those with SCD worsened by low cell dose, low vector copy number, and a return to the SCD phenotype of high erythropoietic stress. These risks may have been mitigated by the use of a plerixafor- mediated stem cell harvest (in place of bone marrow collection) and precollection transfusion therapy; however, the degree to which this will reduce the long-term risk is unknown.
Regarding gene addition, the major concern is the potential risk of an insertion at a promoter site that causes unwanted cellular proliferation or malignant transformation. This issue occurred recently when a patient with cerebral adrenal leukodystrophy developed myelodysplastic syndrome after receiving gene therapy. The viral vector, Lenti-D (Bluebird Bio), differs from those used in SCD-related gene therapy but is similarly designed to add functional copies of a gene into a stem cell. However, based on the location of the LVV insertion, there is concern that it caused the myelodysplastic syndrome (press report, Bluebird Bio Aug 13, 2021). This has not yet been observed (in any LVV-based gene therapy for SCD) but remains a potential risk. Gene editing can also result in unintended modifications at other points along the genome outside of the targeted DNA sequence (off-target effects). Current technology allows us to identify off-target mutations that occur at a high frequency, but it is possible that the more rare ones could (theoretically) escape detection and give cells growth or survival advantages that promote cancer. Further, it is clear that when electroporation is used there is decreased stem cell survival24 ; it is unclear if this will result in any additional negative outcomes.
CLINICAL CASE (continued)
Your patient returns the following week with her family. After several conversations with you (her physician), her family, and her pastor, she decides she understands the risks and benefits of gene therapy and wants to move forward. Your resident asks why it took 3 weeks for her to reach this conclusion.
A sickle cell expert and team familiar with all of the therapeutic options for the treatment of SCD should be required to help an individual with SCD make a significant treatment decision such as gene therapy. The patient should be made aware of the realistic expectations and potential risks of gene therapy, with an emphasis on the importance of shared decision-making between the patient, family, and physician.25 Ample time detailing the known and unknown aspects of gene therapy with the patient and their family will facilitate meaningful discussion and dialogue. It is also important to realize that both benefits and risks are inherent in new procedures, but the potential for cure is also available. Finally, gene therapy still requires myeloablation, which may limit uptake and participation based on both the risks of infertility as well as any baseline organ dysfunction that would make a potential patient ineligible. Thus, thorough deliberation should be undertaken before screening is initiated.
Recent data from gene therapies in progress demonstrate the need for the long-term follow-up and consistent collection of data using common data elements to ensure that outcomes can be compared across trials as well as to the natural history of SCD. Additional data are needed regarding the use of gene therapy for the specific remediation of organ-related complications in SCD as well as in patients whose primary presenting symptom is severe, chronic pain. Finally, enhanced SCD surveillance and longitudinal studies are needed to better understand, quantitate, and compare the potential for malignancy in this population.
Conflict-of-interest disclosure
Julie Kanter: honoraria: Novartis, Graphite Bio, Forma Therapeutics, Agios, Beam Therapeutics, Guidepoint Global, GLG; research funding: National Institutes of Health, National Heart, Lung, and Blood Institute, Health Resources and Services Administration, Centers for Disease Control and Prevention.
Corey Falcon: no competing financial interests to declare.
Off-label drug use
Julie Kanter: nothing to disclose.
Corey Falcon: nothing to disclose.
