
Abstract
Chronic pain in sickle cell disease (SCD) refers to pain present on most days lasting over six months. It can start during childhood and the prevalence increases with age. By adulthood, over 55% of patients experience pain on over 50% of days; 29% reporting pain on 95% of days. The true prevalence of chronic pain in SCD is likely underappreciated as it is mostly managed at home. Patients with chronic pain and SCD frequently seek acute care for exacerbation of underlying chronic pain difficult to distinguish from their usual acute vaso-occlusive crises. When treating chronic pain in SCD, the challenge is distinguishing between non-SCD related etiologies versus chronic pain resulting from SCD pathophysiological processes. This distinction is important to delineate as it will drive appropriate management strategies. Chronic pain in SCD has profound consequences for the patient; is often associated with comorbid psychiatric illnesses (depression and anxiety), not dissimilar from other chronic pain syndromes. They may also experience challenges with sleep hygiene, various somatic symptoms, and chronic fatigue that impair quality of life. How best to treat chronic pain in SCD is not definitively established. Both acute and chronic pain in SCD is typically treated with opioids. Emerging data suggests that chronic opioid therapy (COT) is a suboptimal treatment strategy for chronic pain. This review will discuss the complexity of managing chronic pain in SCD; pain that may be dependent or independent of the underlying SCD diagnosis. We will also describe alternative treatment approaches to high-dose COT.

Learning Objectives
Review common presentations and characterize complex pathophysiology of chronic pain in SCD, recognizing the contribution of non-SCD comorbidities
Identify limitations of and alternatives to chronic opioid therapy (COT) for chronic pain in SCD and the overlap between opioid toxicity and SCD symptoms
Introduction
Chronic pain in sickle cell disease (SCD) refers to pain that is present on most days and has lasted at least 6 months.1 It can start as early as childhood, and its prevalence increases with age. By adulthood, more than 55% of patients experience pain on more than half of days, with nearly one third (29%) reporting pain on 95% of days.2 The true prevalence of chronic pain in SCD is likely underappreciated, because most patients manage their pain at home.2 Patients with SCD and chronic pain frequently seek acute care for an exacerbation of their underlying chronic pain, which is difficult to distinguish from their usual acute vaso-occlusive crises (VOCs). No laboratory markers exist to help define this distinction. When an individual with SCD experiences chronic pain, the challenge is distinguishing the chronic pain that is from non–SCD-related etiologies from chronic pain that results from the SCD pathophysiologic process itself. Understanding and delineating this distinction is critical to determine appropriate disease management strategies.
Chronic pain in SCD has profound consequences for the patient; it is often associated with comorbid psychiatric illnesses such as depression and anxiety, not dissimilar from other chronic pain syndromes.1 Patients with SCD and chronic pain may also experience challenges with sleep hygiene, various somatic symptoms, and chronic fatigue that impair quality of life.1
How best to treat chronic pain in SCD is not definitively established. Both acute and chronic pain in SCD is typically treated with opioids, but emerging data suggest that chronic opioid therapy (COT) is a suboptimal treatment strategy for chronic pain.3 This review will discuss the complexity of managing chronic pain in the patient with SCD; pain that may be dependent or independent of the underlying SCD diagnosis. We will also describe alternative treatment approaches to high-dose COT.
Clinical case 1
A 25-year-old African American woman with hemoglobin (Hb) SS presented to the emergency department (ED) with acute pain. She reports sudden onset of pain in her lower back and thighs, rated 9 of 10 on the numeric pain rating scale. Her pain was not alleviated by her usual home pain regimen of oxycodone 20 mg every 4 to 6 hours as needed. Her outpatient morphine equivalent daily dose (MEDD) is 120 mg, based on prescription fills. Pain was described as throbbing and aching of all joints and thigh muscles that progressed to “pain all over.” She reports chills, sweating, and nausea. She is not currently taking any SCD-modifying therapy and does not have a primary adult hematologist. She receives most of her care via the ED. In the past year, she has had significantly high acute care utilization, with 25 ED visits for pain exacerbations, 20% of which resulted in a hospital admission that has lasted 3 days on average.
Her medical history revealed 1 episode of acute chest syndrome at age 13, a laparoscopic cholecystectomy at age 14, and a normal transcranial Doppler ultrasound screen at age 16. She was lost to hematology follow-up after leaving pediatric care at age 19 when she became pregnant. She struggled with depression as a teenager. She scored a 14 on the Patient Health Questionnaire-9 after the birth of her daughter; she was diagnosed with postpartum depression but declined antidepressant medication and psychotherapy. There are no reports of suicidal ideation or suicide attempts. While in pediatric care, she was prescribed hydroxyurea (maximum tolerated dose of 1500 mg daily) for frequent VOCs with moderate-to-low adherence. Her Hb F peaked at 8%.
In the ED, her vital signs revealed a temperature of 37.3°C, pulse of 110 beats/min, respiratory rate of 12 breaths/min, blood pressure of 129/76 mm Hg, and oxygen saturation of 99% on room air. Physical examination revealed no focal findings, with normal range of motion in all joints. Her hemoglobin level was 8.9 g/dL (at baseline), mean corpuscular volume (MCV) was 83 fL, reticulocyte fraction was 15%, platelet count was 690 000/mm3, and white cell count was 16 800/mm3. Serum electrolytes and kidney function were normal for her age. Chest radiograph showed no infiltrates.
She received intravenous (IV) hydration with NaCl 0.5% at 125 mL/h, 1 dose of IV ketorolac 30 mg, and 3 doses of IV hydromorphone 2 mg every 30 to 60 minutes. She was subsequently admitted for ongoing management of presumed acute VOC, and the hematology team was consulted.
Clinical case 2
A 40-year-old African American man with Hb SC presented for a follow-up hematology visit. He has a 10-year history of daily chronic pain in his low back, knees, and hips. He describes his pain as tingling, aching, and throbbing, with occasional bouts of sharp radiating pain down both legs. His pain intensity fluctuates but at baseline is a 6 of 10 on the numeric pain rating scale and is worst in the mornings. The pain is exacerbated by physical activity, partially relieved by rest, and marginally improved by taking opioid analgesics. Lately, he has had frequent exacerbations of his chronic pain to an intensity of 8 of 10. He presented to the ED 3 times in the last 2 months with acute pain because he ran out of his pain medication early. He reports intermittent swelling of his hands and feet, morning stiffness in all joints, chronic fatigue, feelings of sadness, and difficulty sleeping. He worked in construction but went on disability 6 years ago because of chronic pain. His prescribed home pain regimen includes extended-release morphine sulfate 60 mg 3 times daily and immediate-release morphine sulfate 30 mg up to 6 doses daily. However, he reports taking additional breakthrough doses of his immediate release morphine for pain exacerbations at home; 8 or more doses a day. His daily prescribed outpatient MEDD is 360 mg with no current SCD-modifying therapy.
His medical history includes bilateral hip avascular necrosis (AVN), exaggerated lumbar lordosis, and chronic constipation, with a bowel movement every 3 days.
His vital signs revealed a temperature of 37.6°C, pulse of 69 beats/min, respiratory rate of 10 breaths/min, blood pressure of 139/71 mm Hg, and oxygen saturation of 94% on room air. His weight is 265 lbs, height is 5 ft, 11 inches, and body mass index is 37 kg/m2, which is considered morbidly obese. He has bilateral swelling of knees, feet, and hands. Range of motion evoked pain in all joints. His abdomen is protuberant with soft mobile fecalith masses palpated in his left lower abdomen. Hemoglobin level was 9.0 g/dL (at baseline), MCV was 63 fL, reticulocyte fraction was 4.5%, platelet count was 656 000/mm3, and white cell count was 6100/mm3. Serum electrolyte levels and kidney function were normal for his age. Serum 25-hydroxyvitamin D level was 7 ng/mL. The patient would like to discuss increasing the number of immediate-release morphine sulfate doses prescribed per month so that he can avoid going to the ED.
Causes of chronic pain in SCD
Chronic pain in SCD is a debilitating complication that is associated with increased morbidity and mortality. Pain may be present in 1 or more locations, with or without contributing disease complications such as leg ulcers, AVN, bone infarcts, vertebral body collapse, or any combination thereof.1 SCD complications accumulate over a lifespan; therefore, individuals have an increasing burden of chronic pain with age. Multiple underlying causes of pain can contribute to the evolution of chronic pain in SCD, including repeated acute nociceptive pain from VOCs, inflammatory pain, neuropathic pain, and opioid-induced hyperalgesia (OIH) as depicted in Figure 1, all of which lead to central sensitization (CS).4-7 CS is a condition of hypersensitivity to pain that is associated with both allodynia and hyperalgesia. Allodynia occurs when an individual experiences pain from stimuli that do not normally cause pain. Hyperalgesia occurs when an individual experiences an enhanced sensitivity to a familiar painful trigger.
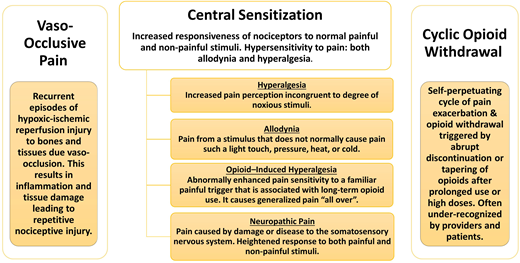
Distinction between 3 different types of pain experienced by patients with SCD.
Distinction between 3 different types of pain experienced by patients with SCD.
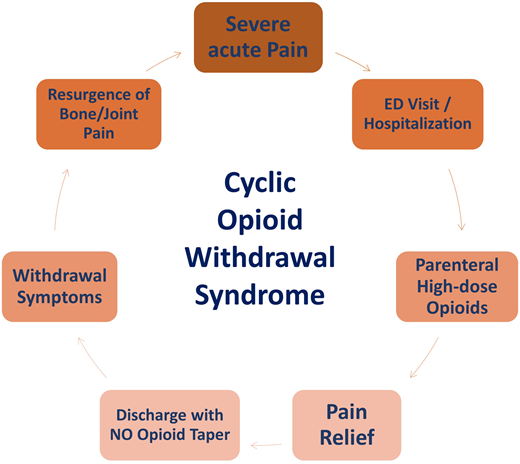
Chronic exposure to high doses of opioids may contribute to chronic pain in SCD from either OIH and/or cyclic opioid withdrawal. Although poorly described in the literature, cyclic opioid withdrawal refers to a pattern of repeated cycles of pain exacerbations along with symptoms of opioid withdrawal. This typically occurs when high doses and/or chronic opioid therapy are abruptly discontinued or when the dose is precipitously decreased after treatment of presumed VOC pain. Effective reductions in plasma opioid levels that can precipitate withdrawal symptoms may occur when there is a change in the administration route, which can lead to a change in the relative potency of the drug; for example, the oral dosing of morphine sulfate needs to be 3 times higher than the parenteral dosing to maintain the same potency. It can also occur when there is a change in opioid potency, which can occur when switching between 2 opioid drugs.
The patient’s clinical course often reveals a gradual increase in multifocal bone, joint, and muscle pain or aching associated with the classic symptoms of opioid withdrawal such as low-grade fevers, chills, nausea, vomiting, diarrhea, abdominal cramps, and depressed mood.8 The pain is often relieved with oral or parenteral opioids, and symptoms of severe acute pain recur once the patient is no longer receiving opioids (Figure 2). Patients with SCD receiving opioids may not be asked about symptoms of withdrawal when they present with worsening pain, and constitutional symptoms that may indicate withdrawal may often be attributed to their usual VOC. As a result, opioid withdrawal in SCD may be grossly underdiagnosed. Failure to treat opioid withdrawal promptly with opioids, via either the oral or parenteral route, and supportive care may lead to dehydration and autonomic distress, resulting in hospitalization for severe pain.
Patients with SCD may also experience a myriad of painful conditions that are unrelated to SCD. Therefore, clinicians should keep an open mind when developing a differential diagnosis for chronic pain in SCD and not assume that all pain in a person with SCD is related to their SCD. Indeed, it may be difficult to discern the true cause of chronic pain because the symptoms of many painful conditions overlap with those of SCD.
Autoimmune diseases and chronic pain
Diagnoses of autoimmune disorders in patients with SCD are often delayed or missed because the symptoms of SCD are similar to those of many autoimmune disorders. Chronic musculoskeletal complications, like AVN, compression spine deformities, and leg ulcers, are not uncommon in SCD. Thus, clinicians may overlook the possibility of a coexisting autoimmune disorder such as rheumatoid arthritis (RA) in patients with chronic pain in SCD.9,10 Although not well documented in the literature, comorbid autoimmune disorders observed in patients with SCD include RA,11 autoimmune hepatitis,12 Crohn’s disease,13 myasthenia gravis,14 and juvenile idiopathic arthritis.15
Clinicians should inquire about a family history of autoimmune disorder and document symptoms that may be suggestive of one, such as alopecia, generalized fatigue, rashes, and pain in multiple joints or involving both large and small joints. If symptoms are suggestive, screening for laboratory markers of autoimmunity, such as antinuclear antibody titers, C-reactive protein, and erythrocyte sedimentation rates, will help guide appropriate rheumatology referral. It is important to note that both erythrocyte sedimentation rate and C-reactive protein may both be elevated in SCD, because they are nonspecific markers of inflammation, and SCD is a chronic inflammatory state. However, if abnormal at baseline, an upward trend may be more indicative of a surrogate marker of rheumatologic disease. Treatment of autoimmune disorders in SCD requires careful orchestration between rheumatology and hematology, because many immunomodulatory agents like steroids and other disease-modifying antirheumatic drugs can induce severe SCD complications such as VOCs or cause cytopenias.16 Therefore, it is important to have a coordinated multidisciplinary team approach to managing patients with both chronic pain in SCD and a comorbid autoimmune disorder.
Neuropathic pain
Neuropathic pain is an increasingly recognized subtype of chronic pain in SCD. This is pain caused by a lesion, medication, or disease of the somatosensory system.17 The somatosensory nervous system allows for the perception of sensations such as touch, temperature, pain, pressure, position, movement, and vibration.18 Individuals with neuropathic pain experience a heightened response to painful and typically nonpainful stimuli - touch, heat, and cold, for example. Patients commonly report sensations of burning, tingling, pins and needles, and numbness.
The lack of a standardized approach for diagnosing neuropathic pain has resulted in unreliable estimates of its prevalence in the general population and among patients with SCD. The prevalence of neuropathic pain in SCD is estimated to be 25% to 40%.19-22 A number of validated screening tools in the form of patient–reported questionnaires can now be used to support a neuropathic pain diagnosis.21,23-25
Because neuropathic pain in SCD is under-recognized, it is also undertreated. Very few patients with SCD are reported as taking neuropathic medications for their chronic pain.20 Examples of medications that have strong supporting evidence as first-line treatment of neuropathic and other pain include tricyclic antidepressants, serotonin noradrenaline reuptake inhibitors, selective serotonin reuptake inhibitors, and gabapentinoids such as gabapentin and pregabalin.26
Psychiatric disorders and chronic pain
Comorbid psychiatric disorders (mood disorders, anxiety, pain catastrophizing, posttraumatic stress disorder, and substance use disorder) are documented in patients with SCD and influence all aspects of the patient experience.29,30 Mood disorders range in spectrum from dysthymia and depressed mood to major depressive episodes and major depressive disorder.
One study reported that more than 35% of adults with SCD had depression,31 a much higher incidence rate than the general population. Among children with SCD, chronic or recurrent pain increases the risk of depression.32 Parent catastrophizing may influence the pain experience of children with SCD33 and significantly contribute to their depressive symptoms.34,35 These studies highlight the bidirectional link between mental illness and pain, with both conditions mutually reinforcing one another.
Opioid therapy has long been associated with depressed mood in persons with chronic noncancer pain. An increasing body of literature suggests that longer duration of opioid therapy of greater than 90 days is associated with a higher incidence of newly reported depressed mood and depressive disorders.36 Similarly, the chronic use of opioids may adversely impact the psychological health of patients with SCD. A recent descriptive study showed that SCD patients on COT reported depression more often than those not on COT5 and self-reported long-acting opioid use in SCD varied as a function of daily negative affect.37 To effectively address chronic pain in SCD, we must also address the mental health of the patient. The American Society of Hematology recently published the 2020 guidelines for sickle cell disease management of acute and chronic pain.38 They suggest that given the high prevalence of psychologic comorbidities that often coexist in the context of pain, routinely screening for depression and anxiety, and targeted screening for other psychologic comorbidities is considered good clinical practice.
COT
Opioids are central to the management of acute SCD pain, and strong evidence supports their use in this setting.39 However, opioids are also used in the management of chronic pain in SCD, despite the lack of evidence supporting their use in treating chronic noncancer pain.40,41 The perceived benefits of COT are often negated by its myriad adverse effects such as CS,42 poor oral health,43 opioid-induced constipation, somnolence, sleep disturbance, cognitive dysfunction, depression, endocrinopathy, tolerance, physiologic opioid dependence, hyperalgesia, and increased overdose- and cardiovascular-associated mortality.44 CS is often mistaken for increased opioid tolerance, causing providers to further escalate opioid dosages in an attempt to ameliorate symptoms. In a study by Karafin et al,45 SCD patients on high doses of opioids had lower health-related quality of life than those on lower doses. Furthermore, patients with chronic pain in SCD on COT had higher pain scores than those who were not on COT.5 Therefore, it is critically important that we explore alternatives to opioids for the management of chronic pain SCD.
Management decisions for case 1: addressing cyclic opioid withdrawal and comorbid mental illness in a young adult with SCD
Initially, this patient’s treatment strategy centered on engaging psychosocial and crisis intervention services. Several barriers to care were identified, including financial difficulty, housing insecurity, transportation challenges, intimate partner violence, and childcare difficulties. Addressing these social determinants became the primary focus of her initial care plan. An extensive psychologic assessment led to a diagnosis of depression, anxiety, and moderate physiologic opioid dependence. Her social worker provided solution-focused therapy and individualized support to encourage her to start an selective serotonin reuptake inhibitor for depression.
The patient was using a standardized opioid risk assessment tool, the patient was identified as being high risk for adverse opioid-related outcomes.46 Without a coordinated multidisciplinary approach to overall disease management, many patients with SCD are inadvertently prescribed opioid doses that far exceed the safety threshold of 90 MEDD recommended by the Centers for Disease Control and Prevention in 2016.47,48 The potential analgesic benefits of a higher MEDD must be carefully weighed against opioid-related toxicity. If pain is not relieved by increasing opioid doses, clinicians should consider clinical explanations such as tolerance, OIH, poorly managed SCD, an unrecognized psychiatric comorbidity, a non–SCD pain–related syndrome, or any combination thereof. The treatment of each of these conditions diverge; higher opioid doses with or without opioid rotation could address physiologic tolerance, whereas lower opioid doses could ameliorate OIH. Cyclic opioid withdrawal should also be considered as a plausible reason for more frequent acute care utilization. Clinicians should be vigilant for signs of withdrawal such as yawning, piloerection, rhinorrhea, increased lacrimation, and diarrhea. Performing an objective assessment of withdrawal using a practice-preferred withdrawal scale, such as the Clinical Opiate Withdrawal Scale (COWS)49 or the Clinical Institute Narcotic Assessment may be beneficial.
At the patient’s most recent ED presentation, an opiate withdrawal assessment was performed using the COWS. Her score of 18 was indicative of moderate opioid withdrawal. This score, her medical history, and her presenting symptoms led to a diagnosis of physiologic opioid dependence and cyclic opioid withdrawal. Transitioning this patient from as-needed oxycodone and frequent IV opioids to sublingual buprenorphine/naloxone may control her symptoms of cyclic opioid withdrawal and physiologic opioid dependence while providing a safer alternative for pain relief. Several buprenorphine preparations are approved for the treatment of acute and chronic pain. Buprenorphine/naloxone is approved for the treatment of opioid use disorder; however, it is increasingly prescribed off-label for chronic pain.50 We used motivational interviewing techniques and shared decision making to transition our patient from daily short acting opioid therapy to buprenorphine/naloxone. Under the guidance of an experienced provider, our patient received her first sublingual dose of buprenorphine/naloxone during an ambulatory clinic visit after overnight abstinence from oxycodone. Her COWS score at induction showed evidence of mild-to-moderate withdrawal, and her induction course followed a modified version of the treatment improvement protocol for buprenorphine induction that was previously reported by McNicholas et al.51
After transitioning to buprenorphine/naloxone, her acute care utilization dropped dramatically, adherence to hydroxyurea improved, and Hb F increased from 8% at baseline to 42% after 12 months. Remarkably, the patient had no ED visits in those 12 months. With ongoing psychosocial support from social work and community liaisons, her Patient Health Questionnaire-9 score gradually improved, and adherence to scheduled outpatient appointments increased by 75%.
Management decisions for case 2: addressing RA, depression, obstructive sleep apnea, vitamin D deficiency, and opioid-induced constipation
Early morning joint stiffness and swelling experienced by this patient were suggestive of an autoimmune disorder. Imaging showed early erosive changes and osteopenia involving radius and ulna with carpal crowding. Laboratory markers that were indicative of an autoimmune disorder include an antinuclear antibody titer of 1:1280 with a speckled pattern, rheumatoid factor positive, C-reactive protein level of 5.5 mg/L, erythrocyte sedimentation rate of 89 mm/h, and anti–cyclic citrullinated peptide antibody titer of 69 U/mL. His low hemoglobin and MCV and elevated platelet count were consistent with anemia of chronic disease superimposed with preexisting hemoglobinopathy. Anemia of chronic disease is common in autoimmune disorders. He was referred to rheumatology and diagnosed with seropositive RA.
A comprehensive psychosocial assessment revealed feelings of hopelessness from multiple comorbid diagnoses. His treatment plan consisted of supportive cognitive behavior therapy, acceptance commitment therapy, and the serotonin noradrenaline reuptake inhibitor duloxetine that also treats neuropathic pain. A physiatry assessment recommended physical therapy to improve joint mobility and function. Vitamin D deficiency (VDD) has been associated with chronic pain in SCD.52 VDD often presents as pain in back, bones, and muscles, low bone density, fatigue, and depressed mood. These symptoms confound the myriad of musculoskeletal and constitutional symptoms that patients with chronic pain in SCD often experience.53 His severe VDD was treated per the American Association of Clinical Endocrinologists guidelines,54 with oral vitamin D3 50 000 IU weekly for 6 weeks. He was subsequently maintained on 5000 IU oral vitamin D3 daily.
An Epworth Sleepiness Scale questionnaire was administered to evaluate the patient’s history of nonrestorative sleep. His score of 12 prompted referral to a sleep specialist, and he was diagnosed with obstructive sleep apnea (OSA). Chronic pain poses limitations on physical activity that can lead to weight gain and worsening OSA symptoms. COT may also contribute to sleep-disordered breathing via suppression of respiratory and sleep centers.44 His treatment plan for OSA included the use of a continuous positive airway pressure machine at night and dietary modifications to support weight loss. Sleep disorders are common among patients with SCD.53 They are associated with increased risk of cardiovascular comorbidities such as higher mean systolic blood pressure, impaired left ventricular diastolic dysfunction, and shorter time to first stroke.53 The combination of nocturnal hypoxemia, increased oxidative stress, increased proinflammatory cytokines, and endothelial dysfunction seen when a patient has both SCD and OSA may potentiate the harmful clinical effects, including increased frequency of pain exacerbations and the associated chronic end-organ damage of SCD.55
The hematology and rheumatology care teams discussed optimal choice of disease-modifying therapy for this patient. Autoimmune disorders may cause pain from the destructive inflammation of multiple joints; therefore, definitive therapy is indicated. Patients with seropositive RA may experience more aggressive pain symptoms and more severe damage to affected joints than with seronegative RA. In addition, the proinflammatory state and ischemic changes from VOCs can exacerbate the bone erosions and cartilage damage associated with RA.56 OSA is often associated with nighttime hypoxia. This may trigger increased sickling and further exacerbate inflammatory pain and VOC; therefore, the underlying SCD must also be meticulously managed. When choosing therapy for concomitant autoimmune disorders in SCD, clinicians must carefully consider the risks of steroid-induced SCD pain exacerbations, and steroid-induced progression of AVN. Caution is warranted with the use of methotrexate and sulfasalazine, because these medications have potential hematologic toxicities.56 The decision was made to treat with a short course of corticosteroids and anti–tumor necrosis factor biologic therapy.
The patient was started on l-glutamine to mitigate ongoing ischemia-mediated VOC from SCD. In a phase 3 trial, oral l-glutamine twice daily was shown to reduce the incidence of pain crises requiring parenteral analgesia.57 l-Glutamine improves nitric oxide bioavailability and mitigates oxidative stress, which could potentially reduce the oxidative damage from RA.56
After initiating RA therapy and SCD-directed therapy with l-glutamine, the patient reported significant improvement in RA-specific symptoms - joint pain, swelling, and stiffness, and successfully tapered his opioids to <90 MME/day within 6 months with and experienced improved bowel motility. He continues to report mild neuropathic pain symptoms that are tolerable.
Discussion
Our cases highlight important concepts for the optimal management of chronic pain in SCD.
A multidisciplinary team approach that addresses the medical and psychosocial drivers of pain is required to adequately treat chronic pain in SCD. The team must include a hematologist and the appropriate subspecialist but, critically, must also include psychosocial and mental health providers, such as a social worker, behavioral health specialist, case manager, and community-based health worker or advocate (Figure 3). This ensures a holistic patient-centered approach is used that adequately addresses social determinants of health in addition to the medical complications that contribute to persistent pain.
Clinicians should keep an open mind when treating chronic pain in SCD and consider a broad differential diagnosis (Figure 4). For each patient, the pathologic basis for chronic pain can evolve; therefore, periodic re-evaluation is warranted, and treatment recommendations should be revised accordingly.
SCD-modifying therapies should be optimized for each patient. The patient in case 1 was treated with hydroxyurea; the patient in case 2 with l-glutamine. Other SCD treatment options for overall disease management should also be considered including chronic hyper-transfusion therapy and the newer agents (crizanlizumab, voxelotor); however, there are limited data available on their efficacy for chronic pain treatment. When appropriate, clinicians should also inform patients of available clinical trials.
We recommend a treatment strategy that tracks MEDD and focuses on harm reduction by transitioning from high-risk opioids to safer options when appropriate. The use of buprenorphine/naloxone is worth considering for patients with SCD on COT who have a high opioid risk score, are experiencing cyclic opioid withdrawal, or meet criteria for physiologic opioid dependence.58 Large studies are needed to further support the use of buprenorphine products for chronic pain in SCD.
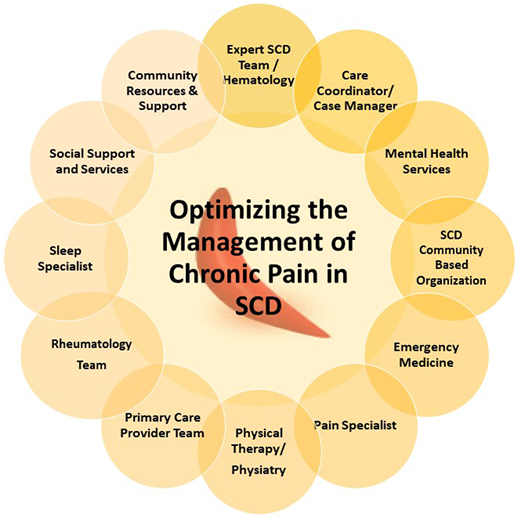
Multidisciplinary team approach to optimizing the management of chronic pain in SCD.
Multidisciplinary team approach to optimizing the management of chronic pain in SCD.

Correspondence
Ifeyinwa Osunkwo, Levine Cancer Institute, Atrium Health, Morehead Medical Dr, 5th Fl LCI-2, Charlotte, NC 28204; e-mail: ify.osunkwo@atriumhealth.org.
