

Abstract
Lower-risk myelodysplastic syndromes (MDS) are characterized by the presence of dysplasia, low bone marrow blast percentage, low number and depth of cytopenia(s), and relatively good-risk karyotpic and molecular abnormalities. A score of ≤3.5 on the Revised International Prognostic Scoring System classifies patients as lower-risk MDS. Information from a mutational profile of the MDS at time of diagnosis (and over serial time points) can be reassuring for predicted behavior of lower-risk MDS compared with one expected to progress more rapidly (higher-risk MDS). Supportive care continues to be the crux of treatment, although the options to reduce transfusion needs have improved in 2020. Erythropoiesis stimulating agents, lenalidomide, and luspatercept address the most frequent (and symptomatic) cytopenia (anemia) and are started only when patients are transfusion dependent. Patients can derive long-term benefits (years) from these approaches but will often progress to higher-risk MDS. Interestingly, some patients with lower-risk MDS can present with an isolated thrombocytopenia for which thrombopoietin receptor analogs such as romiplostim and eltrombopag are options (as long as blast counts are low). The presence of pancytopenia and or intensifying and unremitting clinical symptoms are often treated with hypomethylating agents or (anti–thymocyte globulin if hypocellular MDS is of concern). Targeted therapies are emerging for small subsets of MDS patients with specific somatic mutations (ie, TP53, IDH1/2, FLT3), although currently, there are no approved, mutation-directed medications to treat MDS.
Learning Objectives
Review the treatment paradigm and agents for lower risk MDS, with attention to differing treatment options for transfusion dependent anemia, isolated thrombocytopenia, and pancytopenia
Describe the expected clinical outcomes for patients diagnosed with lower-risk MDS
Describe the impact of somatic mutations on diagnosis of lower-risk MDS (ie; SF3B1mut vs TP53mut) and identify novel therapies under investigation for a targeted precision medicine approach
Clinical case
A 66-year-old man with history of relapsing polychondritis on hydroxychloroquine presented with an isolated macrocytic anemia (hemoglobin, 12.1 g/dL) and minimal symptoms. Flow cytometry for large granular lymphocytic leukemia and paroxysmal nocturnal hemoglobinuria were negative. Other factors were ruled out as contributors to the anemia (hypothyroidism, nutritional deficiencies, infection). Serum erythropoietin (sEPO) level was 77 mIU/mL. A bone marrow biopsy (BMbx) demonstrated a hypercellular marrow (80%) with tri-lineage hematopoiesis, granulocytic hyperplasia, dysmegakaryopoiesis, and 0% blasts consistent with low-grade myelodysplastic syndrome (MDS). Cytogenetic testing revealed 46,XY karyotype. Based on these data, the calculated International Prognostic Scoring System (IPSS) score was 0.5 (low risk), and the IPSS revised (IPSS-R) score was 1 (very low risk), both consistent with a lower-risk MDS. Clinical monitoring was recommended. Approximately 2 years later, he had an acute non-ST elevation myocardial infarction, and he presented to clinic 5 months later. A complete blood count showed worsening anemia (hemoglobin, 8.6 g/dL) and new thrombocytopenia (platelets, 50 000/mL). Repeat BMbx is the same except for 95% cellularity and 1% blasts. NGS testing shows an EZH2 mutation. He is started on the erythropoiesis-stimulating agent (ESA) darbopoetin-alfa (DARBO) at 500 μg every 3 weeks with a meaningful hematologic response.
Introduction
MDS is a heterogenous group of clonal myeloid neoplasms most often characterized by a hypercellular marrow, ineffective hematopoiesis with ≥10% dysplasia in a single cell line, cytopenias, and a risk of progression to acute myeloid leukemia (AML).1 The incidence of MDS in the United States is around 21 000 new cases annually. More than 80% of MDS patients are over 60 years of age,2 and older patients can have a poor long-term overall survival (OS) because of lack of curative therapies (other than hematopoietic stem cell transplant [HSCT], which is limited to a minority of patients). About two-thirds of all MDS patients will present with lower-risk (LR-MDS) disease with minor clinical symptoms and mild cytopenias, and a rare number of patients can have a symptom burden that is out of proportion to their minimally affected laboratory parameters. Notably, for some with LR-MDS, their symptoms can contribute to a decreased quality of life despite their lower-risk scoring, and about 25% of LR-MDS patients will die within 2 years. This clinical heterogeneity has long been recognized.
For all MDS patients, existing scoring systems are used to help identify the risk of MDS progression to AML and to aid in a physician’s treatment recommendations. The IPSS is based on karyotype, bone marrow blast percentage, and number of cytopenias.3 IPSS has since been adopted in clinical trials and subsequently revised in 2012 (IPSS-R), where additional refinement of the specific details on karyotype grouping, degree of cytopenias, and blast counts was made.4 These scoring systems allow for risk stratification into either lower risk (LR) or higher risk. Therapeutic options are guided by these 2 categories and further distinguished by patient-specific characteristics such as age, comorbidities, performance status, and an individual’s goals of care. Nonetheless, these scoring systems currently remain suboptimal because exact attribution and impact on MDS prognosis from the presence of somatic mutational data still remain unclear except for a handful of select genes. For example, LR-MDS patients with a somatic mutation of any of the genes TP53, EZH2, ASXL1, RUNX1, or SRSF2 have a decreased survival than predicted by IPSS.5,6 Bejar et al5 also reported that the presence of EZH2 mutation in combination with the use of LR-IPSS could identify 29% of patients with LR-MDS with a worse prognosis. SF3B1 mutations are associated with a longer OS than calculated by IPSS-R.6,7 Clinical management of the same IPSS-R score is distinct for the aforementioned cases with differing somatic mutational profiles (ie; TP53mut vs SF3B1mut) but as of yet are not formally incorporated to universally adapted scoring systems.8 Importantly, the mere presence of a mutation is not a substitute for the pathologic diagnosis of MDS (ie; requiring the presence of >10% dysplasia) and should not be used as the sole indication for treatment decisions. Mutations in some non-MDS genes indicate the presence of neoplasms that can mimic MDS. These include CALR mutations associated with primary myelofibrosis, C3F3R mutations with atypical chronic myelogenous leukemia and chronic neutrophilic leukemia, and STAT3 mutations with large granular lymphocytic leukemia.
The therapeutic algorithm (Figure 1) for symptomatic LR-MDS is limited to supportive care with transfusions, growth factors, and the 4 US Food and Drug Administration (FDA)-approved drugs in MDS: lenalidomide (LEN), hypomethylating agents (azacitidine or decitabine), and luspatercept. HSCT is the only curative option, but most MDS patients are ineligible because of comorbidities. For those patients that have mild cytopenias with minimal symptoms, watchful observation is appropriate. Early intervention with current modalities has not shown a mortality benefit or impact on clonal evolution in LR-MDS, supporting this strategy.
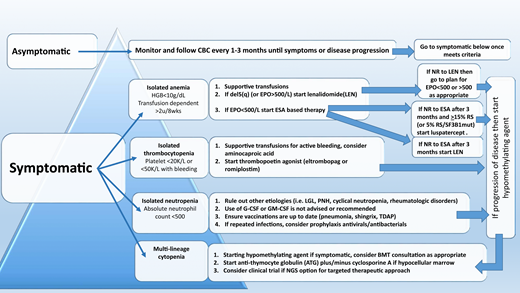
Treatment of anemia
Anemia is the most common symptom in LR-MDS and is present in almost 90% of the cases. With a median age at diagnosis of 71 years, MDS patients can be severely impacted by chronically low levels of hemoglobin, and this can lead to worsening cardiopulmonary function, increased falls, and significant cognitive decline. As such, treatment of anemia is essential for overall health and quality of life. Approach to management is initially focused on packed red blood cell (PRBC) transfusions. However, transfusion-dependent (TD) MDS patients are at higher risk of iron overload and transfusion reactions and report a decreased quality of life. Thoughtful decisions regarding the choice of transfusion support vs initiation of ESA therapy are important, because no evidence supports that early ESA-based therapy improves survival.
A central part of MDS is the ineffective erythropoiesis that contributes to the development of anemia and is characterized by abnormal maturation and differentiation of erythroid progenitors and increased destruction of abnormal erythroblasts.9 The combination of increased proliferation and reduced differentiation results in a net increase of erythroid progenitors and is associated with a stress response that increases EPO levels and signals for preservation of these progenitors in the bone marrow. Unfortunately, there is an uncoupling of proliferation, maturation, differentiation, and resultant increase in cell death, leading to a reduction in functional red blood cells.10 ESAs (recombinant EPO and DARBO) are the first-line agents used for anemia in LR-MDS patients having sEPO levels ≤ 500 U/L and low transfusion burden (Figure 1). The target hemoglobin range for LR-MDS patients is 10 to 12 g/dL with ESA treatment or to achieve an increase in hemoglobin level by ≥1.5 g/dL or a decrease in PRBC transfusion requirements by 4 PRBC transfusions over a period of 8 weeks (Table 1).11 Notably, revised International Working Group (IWG) criteria for assessment of hematologic response have been proposed and are inclusive of a requirement for duration of hematologic improvement to last 16 weeks (vs 8 weeks) among other recommendations.12 In general, overall response rates are 20% to 40% with a general duration of response (DOR) of between 18 and 24 months. Using the validated Nordic scoring system, LR-MDS patients with sEPO < 100 U/L and a transfusion requirement of <2 units of red blood cells (RBCs) have >70% probability of responding to ESA therapy (Figure 2).13 In a phase 3 randomized trial comparing EPO vs best supportive care (BSC), erythroid response rates (RRs) were 36% vs 9.6% at the initial treatment step, which was further increased to 47% in the EPO arm by adding granulocyte colony-stimulating factor and increasing EPO dose in nonresponders.14 Most responding patients had sEPO levels < 200 U/L, and EPO therapy was not associated with OS. In a subsequent phase 3 study of LR-MDS patients with a low transfusion burden, therapy with EPO led to 32% erythroid RR.15 All responses occurred in patients with sEPO < 200 U/L; therefore, approval of ESA use in the European Union was based on this sEPO threshold. In the United States, ESAs are approved for management of chronic anemia, although not specifically for MDS.
Response criteria for hematologic improvement for MDS patients undergoing therapy
| Item | Suggested modified IWG 2018 criteria | IWG 2006 criteria |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
| Item | Suggested modified IWG 2018 criteria | IWG 2006 criteria |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
Abbreviations are as follows: ANC = absolute neutrophil count, Hb = hemoglobin, HTB = high transfusion burden, min = minimum, NTD = not transfusion dependent, pt = patient, PLT = platelet count, TRSFN = transfusion, TB = transfusion burden, RBC = red blood cells, TD = transfusion dependent, TID = transfusion independent, LTB = low transfusion burden.
Only a response duration of at least 16 weeks, however, is considered clinically meaningful.
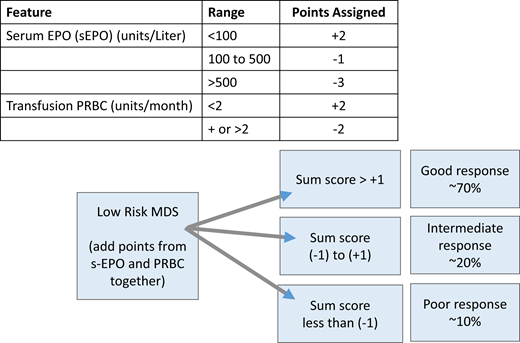
Scoring system for prediction of response to ESA-based therapy in MDS patients. Adapted from Hellström-Lindberg et al.13
Scoring system for prediction of response to ESA-based therapy in MDS patients. Adapted from Hellström-Lindberg et al.13
DARBO has an increased sialylated carbohydrate content that prolongs its half-life and possibly in vivo efficacy. In a phase 2 study of LR-MDS patients with sEPO < 500 U/L, 12-week treatment with DARBO 300 µg/wk resulted in 71% erythroid RR.16 However, in a phase 3 placebo-controlled study of DARBO 500 µg every 3 weeks, RR was lower at 14.7% vs 0% in the placebo group.17 This was likely because of an ineffective dose interval, because RR increased to 34.7% when dose frequency was increased. In an international pooled analysis of 1698 LR-MDS patients treated with ESAs, most responses occurred within 3 months, with a median DOR of 17 months.18 The effect was dose dependent, with EPO 60 000 U/wk and DARBO 300 µg/wk being superior to lower doses. The addition of granulocyte colony-stimulating factor may rescue responses in 10% to 20% of cases, particularly in the presence of ring-sideroblasts (RS).19
Key point
ESA therapy often represents the first step of management for transfusion-dependent LR-MDS patients, with overall response rates of 20% to 40% and an 18- to 24-month duration of response.
MDS progenitors exhibit increased SMAD2/3 signaling that contributes to ineffective erythropoiesis by inhibiting RBC maturation.20 Luspatercept (ACE-536) is a novel recombinant fusion protein, composed of modified activin receptor type IIB linked to the Fc domain of human immunoglobulin. It binds select transforming growth factor β superfamily ligands, decreasing SMAD2/3 signaling and enabling late-stage erythroblast differentiation. In a single-arm phase 2 dose-finding MDS study, 58 LR-MDS patients (hypomethylating agent [HMA] naïve) were treated with luspatercept. Among the patients treated with higher doses of luspatercept, erythroid RR was 63%, with 38% achieving transfusion independence (TI).17 Although low sEPO concentration was predictive of increased response, 43% of patients with sEPO > 500 U/L were still able to attain erythroid response. Most notably, responses were more frequent among patients with SF3B1 mutation compared with non-SF3B1 (77% vs 40%). These findings led to the placebo-controlled phase 3 MEDALIST trial of luspatercept vs placebo in LR-MDS patients with either ≥15% RS or ≥5% RS with SF3B1 mutation, who were TD with disease refractory to or unlikely to respond to ESAs.21 At the dose levels of 1 to 1.75 mg/kg, erythroid RR during the first 24 weeks was 53% in the luspatercept arm vs 12% in the placebo arm. TI for ≥8 weeks was achieved in 37.9% vs 13.2%, respectively (P < .0001). The median DOR was 30.6 weeks in the luspatercept group. The MEDALIST study led to the US FDA approval of luspatercept for MDS with RS and SF3B1 mutation in 2020. The ongoing COMMANDS trial (Efficacy and safety of luspatercept (ACE-536) versus epoetin alfa for the treatment of anemia due to IPSS-R very low, low or intermediate risk MDS in ESA naïve subjects who require red blood cell transfusions.) is a randomized trial comparing luspatercept vs ESA in the upfront setting for TD LR-MDS patients irrespective of SF3B1 status (#NCT03682536). It is likely that combination therapies with luspatercept will be investigated in upcoming clinical trials (ie; with lenalidomide or oral hypomethylating agent).
Key point
Luspatercept was FDA approved in April 2020 for transfusion-dependent MDS with ring sideroblasts. It has been 15 years since the last FDA approval of a drug for the treatment of MDS. Luspatercept was not studied in MDS patients who had prior exposure to LEN or hypomethylating therapy.
The role of iron chelation
MDS patients with high RBC transfusion burden may accumulate excessive amounts of iron and have end-organ damage associated with secondary hemochromatosis. In a phase 2 placebo-controlled study investigating deferasirox in LR-MDS patients with serum ferritin > 1000 ng/mL and transfusion history of 15 to 75 PRBC units, 225 patients were randomized, and iron chelation was associated with a 36.4% risk reduction in event-free survival.22 However, important limitations to highlight in the TELESTO trial (MDS Event Free Survival with Iron Chelation Therapy Study, #NCT00940602) include a long period of enrollment, dramatic reduction in sample size, and a nonstandard definitions of events. Published literature on iron chelation in MDS management is limited by retrospective or single institution studies, which have a lack of statistical prowess. Although chelation can be considered for lower-risk MDS patients with a high transfusion burden along with evidence of end-organ damage from iron deposition, its use should be deliberate and individualized.
LR-MDS with del(5q)
Stemming from the idea of targeting dysregulated immune microenvironment in MDS, early studies investigated the immune-regulatory agent thalidomide in MDS, which resulted in significant toxicity but modest activity in LR-MDS patients.23 This led to the phase 1 MDS-001 study investigating LEN in MDS, which showed manageable toxicity and increased activity in patients with del(5q).24 The phase 2 MDS-003 study tested LEN in 148 TD del5(q) LR-MDS patients. Erythroid RR (per IWG-2000) was 76%, TI was seen in 67%, and 75% experienced a cytogenetic response (50% complete and 25% partial cytogenetic remission).25 Median time to response was 4.6 weeks, and median DOR was 2.2 years.26 The MDS-003 study led to the FDA approval of LEN for del5(q) LR-MDS in 2005. The subsequent phase 3 placebo-controlled MDS-004 study confirmed these results with up to 56% of patients achieving TI for ≥26 weeks.27 Across these studies, the most common side effect of LEN therapy is myelosuppression (50%-60% of patients), which is more pronounced during the first 3 months of therapy. Other less common side effects include rash, diarrhea, pruritus, venous thrombosis, and endocrine pathologies.
Key point
Therapy with LEN is the standard of care for transfusion-dependent del(5q) LR-MDS, with a transfusion independence rate of 67% and a 2- to 3-year duration of response. Patients with concomitant del(5q) and TP53-mutated disease have a decreased likelihood and duration of response.
Given the observed activity of LEN in some MDS patients without del(5q) in the phase 1 study, a phase 2 MDS-002 study was conducted in TD non-del(5q) LR-MDS patients.28 Erythroid RR was 43%, and 26% of patients achieved TI after a median of 4.8 weeks, for which median DOR lasted 41 weeks. The confirmatory phase 3 MDS-005 study also reported 26% TI rate and suggested a more favorable response among patients with baseline sEPO level ≤500 U/L.29 Preclinical data suggest that LEN can restore sensitivity to EPO in MDS cells by stabilizing lipid rafts that are enriched with signaling receptor complexes.30,31 This was further explored in 2 recent phase 3 studies of ESA-refractory TD LR-MDS patients with non-del(5q). In the study by Toma et al,32 combined therapy with LEN-EPO led to higher erythroid RR (39% vs 23%) and TI rate (24% vs 13%) vs LEN monotherapy. However, the DOR was not prolonged (18 vs 15 months, respectively), and the benefit of combination therapy was more prominent in patients with lower transfusion burden and favorable cytogenetics. The E2905 study also investigated this combination with a similar design and reported major erythroid RR of 28.3% in the LEN-EPO arm vs 11.5% LEN monotherapy.33 Among 136 patients who completed 16 weeks of study treatment, RRs were 38.9% vs 15.6% (P = .004). Similar to the first study, the DOR doubled with LEN-EPO vs monotherapy (24 vs 13 months). Concerns regarding the lower than expected RR in the comparator arm (based on prior single arm and randomized trial data) have tempered excitement, although combined therapy can be considered to improve LEN response in non-del5(q) LR-MDS patients.
Key point
Transfusion independence rates are 25% in non-del5(q) transfusion-dependent LR-MDS patients treated with LEN and are associated with a less than 1-year median duration of response. Addition of EPO to Len should be considered and appears helpful to increase frequency of transfusion independence in those EPO-resistant patients with lower EPO level, low transfusion burden, and favorable cytogenetics.
Treatment of thrombocytopenia
Hemorrhage leads to death in 13% of patients with LR-MDS, directly caused by severe thrombocytopenia.34 Intrinsic functional defects of dysplastic megakaryocytes increases bleeding risk, as does concurrent medications frequently used in elderly patients (aspirin, platelet inhibitors, ibuprofen, and others). Platelet transfusions and thrombopoietin-receptor agonists (TPO-RA) are first-line treatment options. Platelet transfusions are not durable, are highly immunogenic, and contribute to splenomegaly. Romiplostim was tested in a randomized phase 2 study of 250 LR-MDS patients treated with subcutaneous dosing vs placebo. Platelet RRs were 36.5% vs 3.6%, respectively, with the incidence of bleeding events and platelet transfusions significantly reduced in the romiplostim group vs placebo (relative risk = 0.71 and 0.35, respectively; P < .0001).35 Although the trial was stopped because of concerns related to excess blasts and progression to AML in the romiplostim arm, 5-year follow-up data did not demonstrate an increased risk of AML or death.36 The oral TPO-RA eltrombopag was also studied in a randomized placebo-controlled phase 2 study of LR-MDS patients.37 Eltrombopag-treated patients had significantly lower bleeding events (14% vs 42%), and higher rate of platelet response (47% vs 3%) compared with placebo (odds ratio, 27.1; 95% confidence interval, 3.5-211.9; P < .0017). Median time to response was 2 weeks. In summary, TPO-RA therapy can improve thrombocytopenia and decrease bleeding in LR-MDS. However, transient elevations of circulating blasts were observed in ∼10% of patients, for which close monitoring is recommended, as well as avoidance of TPO-RA use in MDS patients with excess blasts (>5%).
Key point
Romiplostim increased platelet counts and decreased bleeding events compared with placebo, when given 750 μg subcutaneously once a week. Eltrombopag 150 to 300 mg taken by mouth once daily increased platelet counts and decreased bleeding events compared with placebo in LR-MDS. These agents should be avoided when blast counts are >5% in LR-MDS.
Clinical case (continued)
Our patient experienced an increase in PRBC transfusion requirement at 19 months despite ESA therapy. ESA was stopped. He was reluctant to add LEN given the long-standing history of issues with rashes from relapsing polychondritis. BMbx was performed to evaluate disease status, and it showed 95% hypercellularity with 1% blasts and persistent EZH2 mutation. No RSs were appreciated. Cytopenias progressed to high severity (platelets < 20,000/mL) along with a high transfusion burden for PRBCs. Discussion for next options included azacitidine, clinical trial, and/or HSCT. Given his, young age, profound cytopenias, and high transfusion burden, he was referred for HSCT consultation to initiate typing.
Treatment of multiple and/or refractory cytopenias
Because growth factors have limited efficacy in LR-MDS, patients who are failed by the aforementioned first-line agents are often considered for anti–T-cell immunosuppressive therapy (IST) or HMA therapy. For some patients with pancytopenia, the clinical picture can parallel a bone marrow failure phenotype such as acquired aplastic anemia with a hypocellular marrow (hMDS). Selection of patients who are likely to respond to IST has been challenging because studies are inconsistent about potential predictors of response. Some predictors include younger age, hypocellular marrow, blasts < 5%, normal karyotype, HLA-DR positivity, and short duration of TD.38 A phase 3 trial comparing horse anti–thymocyte globulin (hATG) plus oral cyclosporine (CSA) vs BSC in MDS reported 29% RR with hATG (vs 9% in BSC; P = .02), with a median DOR of 16.4 months.39 In this study, hMDS patients had a much higher RR at 50%, whereas no significant OS or AML-free survival difference was found between the arms. A phase 2 study of single-agent rabbit anti–thymocyte globulin also showed clinical activity with 33% hematologic improvement rate and median DOR of 8.2 months.40 In a large retrospective analysis of 207 MDS patients treated with IST, hATG plus CSA was more effective than rabbit anti–thymocyte globulin, and the highest rate of RBC TI was achieved in patients with hMDS.41 Taken together, the use of IST in hMDS is limited, but hATG plus CSA should be considered. As previously mentioned, younger age can be a predictor of response to IST, so this IST-based therapy should also be considered in younger patients who have LR-MDS without clinical response to ESA-based approaches.
HMA therapy can be used for LR-MDS refractory to first-line therapies. Dose-reduced regimens (ie, 5 days of azacitidine [AZA] 75 mg/m2 per day or 3 days of 50 mg/m2 per day decitabine [DAC]) have been tested in LR-MDS. Two phase 2 trials investigating 5-day AZA combined with EPO in TD LR-MDS after ESA failure reported 20% to 25% erythroid RR and 15% to 20% TI rate.42,43 The limited efficacy observed in these studies is likely because of enrollment of purely anemic patients with significant transfusion burden. Another randomized phase 2 study compared 3-day AZA vs 3-day DAC therapy in LR-MDS and reported superior overall RR (70% vs 49%, P = .03), cytogenetic remission rate (61% vs 25%, P = .02), and OS benefit (20 vs 13 months, P = .1) for the DAC arm.44 However, the difference was likely because of the underdosing in the comparator AZA arm, and a phase 2 study comparing 5 days of AZA vs 3 days of DAC in LR-MDS is ongoing (#NCT01720225). Moreover, most patients enrolled in this study were ESA naïve, which likely accounted for the higher HMA RR compared with other HMA studies done in LR-MDS after ESA failure.
Emerging strategies for management of MDS
A number of emerging therapies with promising RRs are in development and summarized in Table 2. In particular, targeted therapies are of keen interest, especially if toxicities are able to be managed despite combination with other agents.
Emerging therapies for LR-MDS
| Agent | Mechanism of action | Route of administration | Suggested patient population | Single or combination | Response rate | Reference |
|---|---|---|---|---|---|---|
| Roxadustat | Hypoxia-inducible factor (HIF) inhibitor | Oral | LR-MDS (non-del5(q)) with low transfusion burden, sEPO ≤400 U/L | Phase 3 study ongoing ROXA vs Placebo | Dose finding cohort results: N=24. HI-E=54%, TI=38% after 28 wk of treatment. TI=78% at higher dose level 2.5mg/kg | 46 |
| Imetelstat | Telomerase inhibitor | Intravenous | LR-MDS (non del5(q))with high transfusion burden and ESA failure | Phase 2/3 iMERGE study | HI-E=68%, TI for 8 wk=42%, TI for >24 wk=29%, CR=13% and CRi=10%. No PR. High rates of myelosuppression | 47,48 |
| H3B-8800 | Spliceosomal inhibitor: synthetic lethality | Oral | LR-MDS with spliceosome mutations | Phase 1 dose escalation study | 14% HI, no CR/PR PD studies demonstrated dose dependent splicing modulation | 49 |
| APR-246 | TP53 modifier | Intravenous | Treatment naïve HR-MDS/AML with TP53 mutation | Combined with HMA | RR=75-87%, CR=55% CR in phase 2 when combined with HMA | 50,51 |
| Ivosidenib | IDH1 inhibitor | Oral | R/R MDS with IDH1 mutation (N=12) | Phase 1 single agent | CR=5/12 and RR=11/12 | 52 |
| FT-2102 | IDH1 inhibitor | Oral | MDS and AML (N=36) | Phase 1/2 single agent and + HMA | CR/CRi 38% single agent (N=16) CR=27% combo with HMA | 53 |
| Enasidenib | IDH2 inhibitor | Oral | R/R MDS with IDH2 mutation (N=17) | Single or combined | 1/17=CR and 10/17=Response | 54 |
| Agent | Mechanism of action | Route of administration | Suggested patient population | Single or combination | Response rate | Reference |
|---|---|---|---|---|---|---|
| Roxadustat | Hypoxia-inducible factor (HIF) inhibitor | Oral | LR-MDS (non-del5(q)) with low transfusion burden, sEPO ≤400 U/L | Phase 3 study ongoing ROXA vs Placebo | Dose finding cohort results: N=24. HI-E=54%, TI=38% after 28 wk of treatment. TI=78% at higher dose level 2.5mg/kg | 46 |
| Imetelstat | Telomerase inhibitor | Intravenous | LR-MDS (non del5(q))with high transfusion burden and ESA failure | Phase 2/3 iMERGE study | HI-E=68%, TI for 8 wk=42%, TI for >24 wk=29%, CR=13% and CRi=10%. No PR. High rates of myelosuppression | 47,48 |
| H3B-8800 | Spliceosomal inhibitor: synthetic lethality | Oral | LR-MDS with spliceosome mutations | Phase 1 dose escalation study | 14% HI, no CR/PR PD studies demonstrated dose dependent splicing modulation | 49 |
| APR-246 | TP53 modifier | Intravenous | Treatment naïve HR-MDS/AML with TP53 mutation | Combined with HMA | RR=75-87%, CR=55% CR in phase 2 when combined with HMA | 50,51 |
| Ivosidenib | IDH1 inhibitor | Oral | R/R MDS with IDH1 mutation (N=12) | Phase 1 single agent | CR=5/12 and RR=11/12 | 52 |
| FT-2102 | IDH1 inhibitor | Oral | MDS and AML (N=36) | Phase 1/2 single agent and + HMA | CR/CRi 38% single agent (N=16) CR=27% combo with HMA | 53 |
| Enasidenib | IDH2 inhibitor | Oral | R/R MDS with IDH2 mutation (N=17) | Single or combined | 1/17=CR and 10/17=Response | 54 |
CR = complete remission, CRi = complete remission with incomplete count recovery, ESA = erythroid stimulating agent, HI-E = erythroid hematological improvement, HMA = hypomethylating agent, LR = lower risk, PR = partial response, R/R = relapsed/refractory, RR = response rate, sEPO = serum erythropoetin level, TI = transfusion independence.
Clinical case (continued)
The decision was made for treatment with AZA given the transfusion-dependent anemia and progressive thrombocytopenia despite blasts <5%. Absolute neutrophil count was preserved. In conjunction with the HSCT team, recommendations to proceed with HSCT after 2 cycles of AZA was established.
Role of HSCT in LR-MDS
Fit patients with lower-risk IPSS-R with poor-risk genetic features, profound cytopenias, and high transfusion burden are candidates for HSCT.45 Given our enhanced ability to profile MDS at the time of diagnosis (and clonal evolution), we are able to better identify those LR-MDS patients that are at higher risk for progression and allow for earlier curative intent–based therapies.
Conclusions
LR-MDS patients have a notably long survival, and the deliberate selection and sequencing of therapies will lead to an optimal risk/benefit ratio. We continue to identify clonal subpopulations of MDS for which targeted agents can prove useful and potentially restore normal hematopoiesis for some duration. It is likely that these approaches will require insight regarding clonal regression/evolution and the microenvironment in which they are housed. The genetic and biologic heterogeneity of MDS provides significant challenges in developing new clinical therapeutics, which has also been hampered by the lack of good preclinical in vivo models. Nonetheless, the addition of luspatercept to the therapeutic armamentarium for treatment of LR-MDS was encouraging after more than a decade of silence, and excitement regarding novel agents (or combinations of agents) is brightening the horizon.
Correspondence
Hetty E. Carraway, Leukemia Program, Taussig Cancer Institute, Cleveland Clinic, 9500 Euclid Ave, CA60, Cleveland, OH 44195; e-mail: carrawh@ccf.org.
