

Abstract
Heterogeneity is the disease-defining epithet of myelodysplastic syndromes (MDS), a clonal disorder of hematopoietic stem and progenitor cells. During the last decade, significant progress has been made to better understand the diversity of clinical, molecular, cellular, and immunological factors that are bound to the prognosis and outcomes of patients with MDS. Despite the rapid generation of all of this biological information, how to implement it has fallen short. Redefining clinical tools to use this new information remains a challenge. The holistic integration of novel, high-impact individual risk parameters such as patient-reported outcomes or mutational and immunological data into conventional risk stratification systems may further refine patient subgroups, improve predictive power for survival, and provide a next-generation classification and prognosis system for patients with MDS. Dichotomic treatment strategies in patients with MDS according to their patient and disease profiles highlight the importance of precise risk stratification, which may be complemented by the definition of granular cohorts of patients with myeloid neoplasms and a druggable target (ie, IDH1/2 mutations) across conventional blast thresholds.
Learning Objectives
Understand the advantages and limitations of current MDS prognostic scoring systems in patient risk stratification
Gain insight into a potential next-generation classification and prognosis system for patients with MDS
Clinical case 1
A general practitioner referred a 63-year-old man after detecting moderate anemia during a routine peripheral blood analysis. Moreover, the patient had been experiencing moderate fatigue (grade 21 ) since acquiring a viral infection 10 weeks ago. His laboratory values consisted of hemoglobin 8.5 g/dL, absolute neutrophil count 2.5 × 109/L, platelet count 250 × 109/L, serum erythropoietin 148 U/L, and normal liver and kidney values. The results of his cytomegalovirus, Epstein-Barr virus, human herpesvirus 6, and parvovirus B19 polymerase chain reaction diagnostic tests were negative, and so far, he had not received packed red blood cell (pRBC) transfusions.
Subsequent bone marrow aspiration revealed isolated erythroid dysplasia in >20% of cell lines, 24% ring sideroblasts (RSs), and 3% marrow blasts, consistent with the diagnosis of myelodysplastic syndrome (MDS) with RSs and single-lineage dysplasia (MDS-RS-SLD). Karyotype analysis confirmed trisomy 19, and molecular studies of the bone marrow aspirate detected TET2 (35% variant allele frequency [VAF]), DNMT3A (VAF, 25%), and SF3B1 (VAF, 22%) mutations.
Brief overview of MDSs
MDSs are very heterogeneous clonal disorders of hematopoietic stem and progenitor cells and are usually suspected if a (mostly elderly) patient presents with unexplained cytopenia in routine peripheral blood analysis.2-4 The extent of clinical presentation can vary significantly; the spectrum ranges from a mild disease course with minimal or no intervention needed to patients with multiple treatment failures and early progression to acute myeloid leukemia (AML). The highly variable clinical course of patients with MDS represents a challenge, not only with regard to individual prognosis assessment but also in terms of the decision about appropriate but still limited treatment regimens.5-7
Clinical case 1 (continued)
The diagnosis of MDS-RS-SLD is confirmed, but what’s next?
The manual count of bone marrow blasts, 3% in our patient’s case, is fundamental for risk assessment. It is important to note that the percentage of blast counts matters with regard to prognosis, even below the 5% threshold. Therefore, bone marrow smears should be assessed by an experienced hematopathologist. In addition, cytogenetic analysis helps in predicting risk and selecting the right individual treatment strategy. Because of the highly variable prognosis of patients with MDS, prognostic systems allowing risk stratification and subsequent therapeutic decision are of utmost importance. On the basis of the International Prognostic Scoring System (IPSS),8 the patient has a score of 0.5 (intermediate 1 risk), whereas with the revised IPSS (IPSS-R),9 the patient’s score is 4.0 (intermediate).
After initial diagnosis, the patient underwent first-line therapy with an erythropoiesis-stimulating agent (ESA). Nevertheless, after 12 months, his anemia worsened, and he required RBC transfusions (RBC transfusion dependent [RBC-TD]) every 1 to 2 months. Thus, after ESA failure and recent U.S. Food and Drug Administration and European Medicines Agency (EMA) approval of luspatercept, the patient is now eligible for luspatercept treatment.10,11
Current classification of risk and prognosis
MDS prognostic scoring systems have been the focus of research for many years.12 The most commonly used include the IPSS,8 revised IPSS (IPSS-R),9 World Health Organization (WHO) Prognostic Scoring System,13 and MD Anderson Prognostic Scoring System.4,12,14,15
Since 1997, the IPSS8 has been a widely accepted standard for assessing prognosis and stratification of primary untreated adult patients with MDS. The IPSS includes covariates for prognostic discrimination such as the number of cytopenias at initial diagnosis, the percentage of bone marrow blasts, and the number of cytogenetic abnormalities (Tables 1 and 2),8 which distinguish four prognostic categories (low-, intermediate 1–, intermediate 2–, and high-risk disease) with significant differences in overall survival (OS) and rate of AML transformation.8,14 Until today, this simple and highly reproducible prognostic scoring system has been of essential importance and has been the basis for a number of MDS-specific drug approvals within the last 2 decades.12,14,16 However, several limitations of the IPSS became evident, most likely due to the fact that only little weight is given to the diversity of cytogenetic changes and to the extent of cytopenias in mainly patients with RBC-TD anemia.17
IPSS8
| Score | |||||
|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 1.5 | 2.0 | |
| Medullary blasts, % | 0-4 | 5-10 | — | 11-20 | 21-29 |
| Number of cytopenias* | 0-1 | 2-3 | — | — | — |
| Cytogenetic risk group† | Low | Intermediate | High | — | — |
| Score | |||||
|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 1.5 | 2.0 | |
| Medullary blasts, % | 0-4 | 5-10 | — | 11-20 | 21-29 |
| Number of cytopenias* | 0-1 | 2-3 | — | — | — |
| Cytogenetic risk group† | Low | Intermediate | High | — | — |
Low risk = normal karyotype, 5q-, 20q-, -Y; intermediate risk = all other aberrations; High risk = complex karyotype (≥3 anomalies), chromosome 7 anomalies.
Platelets <100 000/μL; hemoglobin <10 g/dL, absolute neutrophil count <1 800/μL.
IPSS prognostic risk categories8
| Score | Risk groups |
|---|---|
| 0 | Low risk |
| 0.5-1 | Intermediate risk 1 |
| 1.5-2 | Intermediate risk 2 |
| ≥2.5 | High risk |
| Score | Risk groups |
|---|---|
| 0 | Low risk |
| 0.5-1 | Intermediate risk 1 |
| 1.5-2 | Intermediate risk 2 |
| ≥2.5 | High risk |
To refine the IPSS, multiple statistically weighted clinical and genetic features were integrated to generate a new prognostic categorization model.9,17 Since 2012, the IPSS-R (Tables 3 and 4) has been the standard tool to assess the risk of disease progression and death of patients newly diagnosed with MDS. The model captures additional and more precise prognostic elements (eg, chromosomal abnormalities, percentage blast count, severity of cytopenia) and defines five rather than four major prognostic categories (very low, low, intermediate, high, and very high risk).9
IPSS-R9
| Score | |||||||
|---|---|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 1.5 | 2 | 3 | 4 | |
| Cytogenetic group* | Very good | — | Good | — | Intermediate | Poor | Very poor |
| Medullary blasts, % | ≤2 | — | >2 to <5 | — | 5-10 | >10 | — |
| Hemoglobin | ≥10 | — | 8 to <10 | <8 | — | — | — |
| Platelets | ≥100 | 50 to <100 | <50 | — | — | — | — |
| ANC | ≥0.8 | <0.8 | — | — | — | — | — |
| Score | |||||||
|---|---|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 1.5 | 2 | 3 | 4 | |
| Cytogenetic group* | Very good | — | Good | — | Intermediate | Poor | Very poor |
| Medullary blasts, % | ≤2 | — | >2 to <5 | — | 5-10 | >10 | — |
| Hemoglobin | ≥10 | — | 8 to <10 | <8 | — | — | — |
| Platelets | ≥100 | 50 to <100 | <50 | — | — | — | — |
| ANC | ≥0.8 | <0.8 | — | — | — | — | — |
ANC, absolute neutrophil count.
Very good = del(11q), -Y; good = normal karyotype, del(20q), del(5q), del(12p), double including del(5q); intermediate = +8, del(7q), i(17q), +19, any other single or double independent clone; poor = −7, inv(3)/t(3q)/del(3q), double including −7/del(7q), complex: 3 abnormalities; very poor = complex >3 abnormalities.
IPSS-R prognostic risk categories and clinical outcomes9
| Score | Risk groups | Median survival, y | Median time to 25% AML evolution, y |
|---|---|---|---|
| 0-1.5 | Very low risk | 8.8 | Not reached |
| 1.5-3 | Low | 5.3 | 10.8 |
| >3-4.5 | Intermediate | 3.0 | 3.2 |
| 4.5-6 | High | 1.6 | 1.4 |
| >6 | Very high | 0.8 | 0.73 |
| Score | Risk groups | Median survival, y | Median time to 25% AML evolution, y |
|---|---|---|---|
| 0-1.5 | Very low risk | 8.8 | Not reached |
| 1.5-3 | Low | 5.3 | 10.8 |
| >3-4.5 | Intermediate | 3.0 | 3.2 |
| 4.5-6 | High | 1.6 | 1.4 |
| >6 | Very high | 0.8 | 0.73 |
Chronic myelomonocytic leukemia is a myelodysplastic/myeloproliferative neoplasm with a highly variable clinical course and prognosis. The chronic myelomonocytic leukemia–specific prognostic scoring system (CPSS) groups patients into risk categories by accounting for cytogenetic abnormalities, disease subtype according to French-American-British and WHO classifications, and RBC transfusion dependency. The CPSS stratifies patients into 4 different risk groups with significantly different survival and risk of AML evolution.18,19 Recently, the CPSS model was updated to include molecular abnormalities, including ASXL1, RUNX1, NRAS, and SETBP1 mutations.19
In patients with lower-risk (LR) MDS (IPSS-R very low to intermediate risk up to 3.5 points20 ), therapy is aimed mainly at improving cytopenia(s) to prevent complications such as bleeding and severe infections, decreasing transfusion burden, and improving quality of life.2 Conversely, in patients with higher-risk (HR) MDS (IPSS-R intermediate risk above 3.5 points,20 high, or very high risk), a more aggressive treatment strategy, including hypomethylating agents (HMAs) or allogeneic hematopoietic stem cell transplant (allo-HCT) with the aims of delaying disease progression, improving survival rates, and potentially curing the disease, should be initiated.2,5 These dichotomic treatment strategies in patients with LR- vs HR-MDS highlight the importance of precise risk stratification at initial diagnosis. Because age did not affect AML transformation risk, the patient’s age is not formally included in the IPSS or IPSS-R, and individual survival prediction based on age and risk status is realized by the age-adjusted IPSS-R (IPSS-RA) formula: ([Age in years − 70] × (0.05 − [IPSS-R risk score × 0.005]).9,21
Our 63-year-old patient in clinical case 1 has an age-adjusted IPSS-RA score of 3.79 (intermediate). We recommend adjustment by age especially in patients with MDS younger than 50 years old, when a potentially curative allo-HCT is considered a possible treatment option.
In some cases, adding age to IPSS-R could potentially upstage patients in the intermediate-risk group to HR disease (IPSS-RA score, >3.5). Patients with an intermediate-risk IPSS-R score represent a group with a highly divergent clinical outcome due to widely variable disease courses.22 Age ≥66 years, peripheral blood blasts ≥2%, and history of RBC transfusion have been identified as additional stratification factors for this challenging subgroup of patients with MDS.22 These factors, all of them associated with inferior survival, enable the classification of patients with IPSS-R intermediate-risk MDS into 2 prognostic subgroups (intermediate-favorable vs intermediate-adverse) with significant divergent outcomes.22 In addition, outcomes of IPSS-R intermediate-risk patients and those with confirmed SF3B1 mutation may be underestimated because of the favorable prognosis of SF3B1-positive patients.23 However, other molecular data, such as TP53 and EZH2, can upstage patients from intermediate-risk into HR category.24,25
Previous analyses demonstrated that comorbidities also had a significant independent impact on survival.26 In fact, the addition of comorbidity scores such as the MDS-specific comorbidity index27 or the newly developed MDS-specific frailty index28 to prognostic systems such as IPSS and IPSS-R can further enhance their prognostic value.26,29,30 Nevertheless, their standardized incorporation into daily workup has not become reality in many centers.
Apart from comorbidities, fatigue is an important marker in individual risk assessment. Independent from current standard risk classification systems, self-reported fatigue severity is a negative prognostic factor for survival.31 A recent international observational study compared the ability of IPSS and IPSS-R to capture baseline fatigue burden in patients with newly diagnosed MDS.32 After stratifying patients according to IPSS score, there was a lack of sensitivity in capturing the burden of fatigue across its four risk categories.32 Contrarily, the IPSS-R determined clearly distinct subgroups with regard to burden of fatigue and thus provided a better stratification of patients related to fatigue severity, possibly enhancing patient management in clinical practice.32 Because fatigue is a readout of several factors, including age, inflammation, pain, emotional distress, sleep disturbance, anemia, and diminished activity level, its regular assessment is recommended in clinical decision making by completing fatigue questionnaires at baseline and during the course of therapy.31,32
Clinical case 2
Another 63-year-old man also presented initially with moderate anemia (hemoglobin, 7.9 g/dL) but normal absolute neutrophil and platelet counts. His serum erythropoietin level was 122 U/L, and he had not received RBC transfusions. A subsequent bone marrow assessment confirmed the diagnosis of MDS-RS-SLD with 2% bone marrow blasts and 21% RSs. Cytogenetic analysis showed trisomy 19, and molecular studies of the bone marrow aspirate confirmed high-risk somatic mutations, including ASXL1 (VAF, 28%), ETV6 (VAF, 32%), and U2AF1 (VAF, 22%). Similar to the patient in clinical case 1, this patient has an IPSS score of 0.5 (intermediate 1 risk), but an IPSS-R score of 3.5 (intermediate). In accordance with the current treatment guidelines, the patient underwent first-line therapy with an ESA. Nevertheless, after 10 weeks of ESA treatment, his hemoglobin levels dropped from 7.9 g/dL initially to 6.2 g/dL, and he became heavily RBC transfusion dependent (6 to 8 RBCs units/month). A subsequent bone marrow diagnostic test demonstrated slightly rising blast counts (4% bone marrow blasts) with 32% RSs. Similar to the patient in clinical case 1, he is now eligible for luspatercept treatment after ESA failure.
Does the molecular coat fit? The impact of SF3B1 mutations
The compendium of common MDS-associated somatic mutations has extensive implications in patient care and prognosis,33 but single (or even cluster) gene mutations are not incorporated into current prognostic scoring systems.4,14 Our patient in clinical case 1 has an IPSS score of 0.5 (intermediate 1 risk; LR-MDS) and an IPSS-R score of 4.0 (intermediate; HR-MDS). The discrepancy between IPSS and IPSS-R can result in a potential therapeutic dilemma. In such a clinical case, the individual molecular profile can be crucial in terms of treatment decision and with regard to the question whether allo-HCT should be considered.
The molecular abnormalities described in clinical cases 1 and 2 demonstrate in an exemplary way how the patient’s individual prognosis depends on the predominant mutations. The 2 clinical cases display similarities in laboratory values, bone marrow blast counts, karyotypes, and risk classification scores (Table 5), but the patient in clinical case 1 exhibits somatic mutations associated with a rather favorable outcome (TET2, DNMT3A, SF3B1),23,34 whereas those in the patient in clinical case 2 are linked to an adverse prognosis (ASXL1, ETV6, U2AF1).34,35
Comparison of clinical variables at initial diagnosis
| Clinical case 1 | Clinical case 2 | |
|---|---|---|
| Hb level | 8.5 g/dL | 7.9 g/dL |
| PLT level | 250 × 109/L | 192 × 109/L |
| ANC count | 2.5 × 109/L | 2.7 × 109/L |
| BM blast count | 3% | 2% |
| Cytogenetic analysis | +19 | +19 |
| Molecular analysis | TET2 (VAF, 35%) | ASXL1 (VAF, 28%) |
| DNMT3A (VAF, 25%) | ETV6 (VAF, 32%) | |
| SF3B1 (VAF, 22%) | U2AF1 (VAF, 22%) | |
| MDS WHO subtype | MDS-RS-SLD | MDS-RS-SLD |
| IPSS | 0.5 (intermediate 1) | 0.5 (intermediate 1) |
| IPSS-R | 4.0 (intermediate) | 3.5 (intermediate) |
| Clinical case 1 | Clinical case 2 | |
|---|---|---|
| Hb level | 8.5 g/dL | 7.9 g/dL |
| PLT level | 250 × 109/L | 192 × 109/L |
| ANC count | 2.5 × 109/L | 2.7 × 109/L |
| BM blast count | 3% | 2% |
| Cytogenetic analysis | +19 | +19 |
| Molecular analysis | TET2 (VAF, 35%) | ASXL1 (VAF, 28%) |
| DNMT3A (VAF, 25%) | ETV6 (VAF, 32%) | |
| SF3B1 (VAF, 22%) | U2AF1 (VAF, 22%) | |
| MDS WHO subtype | MDS-RS-SLD | MDS-RS-SLD |
| IPSS | 0.5 (intermediate 1) | 0.5 (intermediate 1) |
| IPSS-R | 4.0 (intermediate) | 3.5 (intermediate) |
ANC, absolute neutrophil count; BM, bone marrow; Hb, hemoglobin; PLT, platelet.
This molecular discrepancy in both cases is associated with completely different individual risks of disease progression and survival. It becomes clear that the currently available prognostic scoring systems are not sufficient to derive personalized and precise therapeutic decisions in the absence of data on molecular abnormalities. Thus, the addition of molecular data to the IPSS-R will be essential to create a personalized, dynamic risk prediction model with the ability to make individual treatment recommendations (Figure 1, Table 6). The development of such a new molecular IPSS-R system is currently in progress.
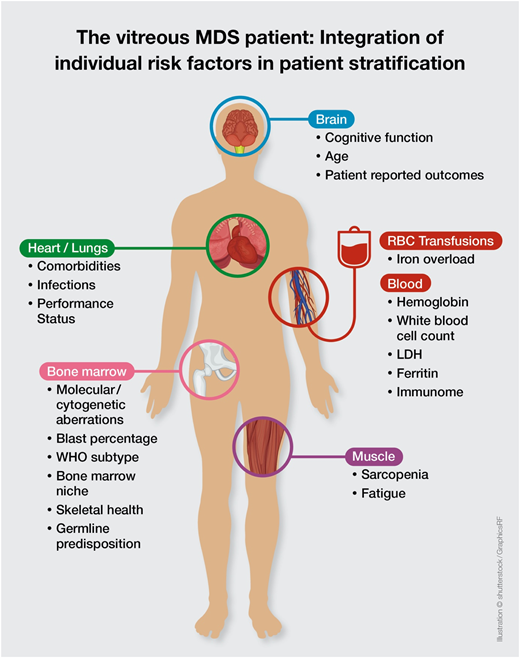
The vitreous MDS patient: integration of individual risk factors in patient stratification.
The vitreous MDS patient: integration of individual risk factors in patient stratification.
Key factors for the development of a potential personalized medicine approach60
| Variable | Entity | Grading | Potential clinical consequence |
|---|---|---|---|
| Performance status | ECOG 0-1 | Good | Standard therapy including allo-HCT |
| ECOG >1 | Poor | Supportive care or low-intensity therapy | |
| EPO level | <200 U/L | Low | Treatment with ESA in case of anemia |
| >200 U/L | High | No ESA (limited response to ESA) | |
| Ferritin level | >1500 ng/mL | High | Treatment with iron chelation* |
| Genetics | del(5q) | Targeted treatment with lenalidomide* | |
| Normal karyotype, 20q-, -Y: and absence of poor-risk molecular abnormalities | Good risk | Standard therapy or supportive care only | |
| All other aberrations, including complex karyotype, or poor-risk molecular abnormalities | Poor risk | Intensified surveillance strategy, allo-HCT, clinical trial | |
| Druggable molecular targets | SF3B1 mutation | Treatment with luspatercept* | |
| TP53 mutation | Treatment with TP53 modulators | ||
| TP53 WT | Treatment with Nutlins | ||
| IDH1 mutation | Treatment with IDH1 inhibitors | ||
| IDH2 mutation | Treatment with IDH2 inhibitors | ||
| Spliceosome mutations | Treatment with spliceosome modulators | ||
| Prognostic scoring systems (eg, IPSS-R) | IPSS-R score ≤3.5 | Good risk | Standard therapy or supportive care only |
| IPSS score >3.5 | Poor risk | Hypomethylating agents,* allo-HCT | |
| Inflammatory signature | Yes | Anti-inflammatory treatment | |
| No | Standard therapy |
| Variable | Entity | Grading | Potential clinical consequence |
|---|---|---|---|
| Performance status | ECOG 0-1 | Good | Standard therapy including allo-HCT |
| ECOG >1 | Poor | Supportive care or low-intensity therapy | |
| EPO level | <200 U/L | Low | Treatment with ESA in case of anemia |
| >200 U/L | High | No ESA (limited response to ESA) | |
| Ferritin level | >1500 ng/mL | High | Treatment with iron chelation* |
| Genetics | del(5q) | Targeted treatment with lenalidomide* | |
| Normal karyotype, 20q-, -Y: and absence of poor-risk molecular abnormalities | Good risk | Standard therapy or supportive care only | |
| All other aberrations, including complex karyotype, or poor-risk molecular abnormalities | Poor risk | Intensified surveillance strategy, allo-HCT, clinical trial | |
| Druggable molecular targets | SF3B1 mutation | Treatment with luspatercept* | |
| TP53 mutation | Treatment with TP53 modulators | ||
| TP53 WT | Treatment with Nutlins | ||
| IDH1 mutation | Treatment with IDH1 inhibitors | ||
| IDH2 mutation | Treatment with IDH2 inhibitors | ||
| Spliceosome mutations | Treatment with spliceosome modulators | ||
| Prognostic scoring systems (eg, IPSS-R) | IPSS-R score ≤3.5 | Good risk | Standard therapy or supportive care only |
| IPSS score >3.5 | Poor risk | Hypomethylating agents,* allo-HCT | |
| Inflammatory signature | Yes | Anti-inflammatory treatment | |
| No | Standard therapy |
ECOG, Eastern Cooperative Oncology Group; EPO, erythropoietin; WT, wild type.
FDA approved
If we go back to our clinical cases 1 and 2, it can be concluded that both patients display RSs ≥15%, but only the patient in clinical case 1 carries a mutation of SF3B1. In MDS with RSs, SF3B1 mutations define a homogeneous subgroup with isolated erythroid dysplasia and favorable prognosis.34 Patients with MDS with RSs and wild-type SF3B1 (clinical case 2) are characterized mainly by multilineage dysplasia and an unfavorable prognosis.34
Within the phase 2 PACE-MDS study, luspatercept (ACE-536), an ActRIIB ligand trap fusion protein, was investigated in anemic patients with LR-MDS.11 Among patients treated with luspatercept, 29 (69%) of 42 RS-positive patients achieved erythroid hematologic improvement vs only 3 (43%) of 7 RS-negative patients. Among SF3B1-positive patients, 77% (24 of 31) achieved erythroid hematologic improvement compared with 40% (6 of 15) of SF3B1-negative patients.11 Within the subsequent placebo-controlled phase 3 MEDALIST trial, the safety and efficacy of luspatercept in TD patients with LR-MDS and RSs (with either ≥15% RSs or ≥5% RSs if SF3B1-positive), who were either refractory to or unlikely to respond to ESA, were confirmed.10 Of 153 patients receiving luspatercept, 93% had an SF3B1 mutation, and 38% achieved the primary endpoint of RBC transfusion independence for 8 weeks or longer compared with 13% receiving placebo (P < .001).10 Interestingly, the percentages of patients with a response to luspatercept treatment were similar, regardless of SF3B1 allelic burden and the total number of baseline somatic mutations.10
After the recent U.S. Food and Drug Administration and European Medicines Agency approvals of luspatercept, both of our patients are now eligible to receive luspatercept treatment. However, because of the existing HR mutational profile (ASXL1, ETV6, U2AF1), including the absence of SF3B1 mutation in the patient in clinical case 2, an intensified surveillance strategy to detect disease progression early should be considered.
Are there other potential treatment options available after ESA failure?
When restricted to primary endpoint responders RBC- transfusion independent (RBC-TI, ≥8 weeks) with an RBC transfusion burden similar to that in the luspatercept trial (≥2 units/8 weeks), the response rate of lenalidomide was almost comparable to that of luspatercept36 (26.9% vs 38%, respectively) in a phase 3, randomized, placebo-controlled study in LR-MDS non-del(5q) patients ineligible for or refractory to ESA treatment36 . However, lenalidomide is not registered for that indication, and grade 3 or 4 neutropenia and thrombocytopenia were reported in 61.9% and 35.6% of lenalidomide-treated patients, respectively.36 In a randomized phase 3 trial comparing lenalidomide monotherapy with lenalidomide plus ESA in ESA-resistant, RBC-TD (≥4 units/8 weeks) patients with LR-MDS, the overall rate of RBC-TI ≥8 weeks was 13.8% vs 24.2% in the lenalidomide vs lenalidomide plus ESA arms, respectively.37
Moreover, in a phase 2 study evaluating azacitidine with or without ESA in patients with ESA-resistant RBC-TD (≥4 units/8 weeks) LR-MDS, RBC-TI ≥8 weeks was achieved in 14.3% of patients receiving azacitidine plus ESA and in 16.3% receiving azacitidine monotherapy.38 These results were consistent with those of the Nordic MDS group, who found that RBC-TI was achieved in 20% of cases in a similar patient population (LR-MDS with RBC-TD ≥4 units/8 weeks and ESA resistance) treated with azacitidine monotherapy, but response duration was short at <6 months in most patients.39 In these 2 azacitidine trials, specific results for patients with RS after ESA failure were not available. Therefore, treatment with luspatercept remains our preferred option in our 2 clinical cases.
Incorporation of molecular data: curse or blessing?
Generally, the prognostic significance of somatic mutations in patients with MDS is connected to other risk factors, including karyotype, blast number, and cytopenias (Table 6), which are captured by existing clinical risk scoring systems such as the IPSS-R. Genome sequencing of 104 genes in a study of 944 patients with MDS showed that 25 of 48 mutations were associated with reduced OS, including RUNX1, ASXL1, NPM1, EZH2, TP53, PRPF8, LUC7L2, NRAS, KRAS, FLT3, PTPN11, NF1, LAMB4, GATA2, SMC1A, and STAG2.33,40 Again, only SF3B1mut status was associated with improved OS40 ; even after adjustment for IPSS-R risk groups, the mutation was strongly associated with a favorable impact on OS in patients with <5% bone marrow blasts.13,16,33 Contrarily, SRSF2, ASXL1, and U2AF1 mutations had an independent negative impact on OS in patients with blast percentages <5%, but not in patients with higher blast percentages (clinical case 2).16,33 Thus, the prognostic significance of MDS-associated mutations should always be considered with regard to the individual bone marrow blast count. Interestingly, the same pattern has been described for the impact of RBC-TD, where its negative impact was rather moderate in MDS with excess blasts.13
In a comprehensive genetic analysis with the aim to identify an association between somatic mutations and clinical variables (OS, severity of cytopenia, proportion of blasts), TP53, EZH2, ETV6, RUNX1, or ASXL1 mutations were associated with an OS similar to that of patients in the next-higher IPSS risk group.41 Mutations of IDH2 were associated with shorter OS, whereas IDH1 mutations were only slightly associated with reduced OS, demonstrating the diverse influence of these strongly related genes.33
Although there is broad consensus on the impact of mutations on prognosis, often there is no clinical consequence, given the paucity of available treatment options. This is a currently unmet medical need that requires novel therapeutic approaches.
Clinical case 3
A 68-year-old woman with known MDS with isolated del(5q) according to WHO 2016 criteria42 and confirmed TET2 (VAF, 25%) and DNMT3A (VAF, 18%) somatic mutations has been receiving lenalidomide treatment for the past 25 months because of RBC-TD. Eight weeks ago, bone marrow diagnostic tests confirmed sustained complete cytogenetic remission, but she now presents with new RBC-TD anemia (hemoglobin, 6.5 g/dL) and worsening thrombocytopenia (platelet count, 48 × 109/L). A subsequent bone marrow assessment showed a rising blast count of 12%. In the setting of suspected disease progression or treatment failure, we recommend repetition of next-generation sequencing testing. Molecular studies revealed a new TP53 mutation (VAF, 30%) with multihit allelic state and slightly rising VAFs for TET2 (VAF, 38%) and DNMT3A (VAF, 32%) mutations. Furthermore, cytogenetic analysis revealed reemergence of del(5q) plus a 17p deletion in 8% of metaphases.
How does TP53 matter?
First, the presence of a TP53 mutation (in ∼10% to 15% of MDS cases) significantly affects the prognosis of patients with MDS.43 TP53 mutations are selectively enriched in cases with complex karyotype (∼70%)44 or therapy-related myeloid neoplasms, and the mutation independently predicts poor OS even after correcting for clinical variables.45 Especially patients with TP53 mutations and complex karyotypes represent a group with an extremely poor prognosis and an OS <6 months.
Moreover, TP53 mutational burden seems to matter with regard to clinical outcome and stratifies distinct prognostic groups independently of clinical prognostic scoring systems.46 In a retrospective study of 219 patients with MDS, patients with a TP53 VAF >40% had a median OS of 124 days; the same OS was not reached in patients with VAF <20% (P < .01).46 In patients with LR-MDS, evaluation of OS determined a TP53 VAF threshold of 6% as an optimal cutoff for patient stratification.47 No significant impact on PFS or OS was observed in patients with LR-MDS with a VAF <6%, who remained stable for long periods without progression.47
Bernard et al48 recently published data about the important prognostic role of TP53 allelic state. In a cohort of 3324 patients with MDS, 490 TP53 mutations in 380 patients (11% of the cohort) were characterized. Analysis revealed a segregation of TP53 into 2 states: a monoallelic state (one wild-type allele remaining) was detected in 33% of patients with TP53 mutations, and a multihit state (TP53 altered multiple times) was shown in 67% of patients with TP53 mutations.48 Interestingly, the allelic state of TP53 was associated with clinical presentation and patient outcome. Monoallelic TP53 state was linked to a more favorable disease, including less severe cytopenias, lower bone marrow blast counts (4% vs 9%), and enrichment in LR-MDS subtypes compared with patients with multihit TP53 state.48 Moreover, patients with monoallelic TP53 state had rather similar survival rates compared with patients with wild-type TP53 in accordance with their IPSS-R stratification.48
On the contrary, multihit allelic TP53 state including a del17p, as in our patient in clinical case 3, was associated with complex karyotypes, worse OS, adverse prognostic subgroups independent of the IPSS-R, and higher rates of AML progression.48 Thus, TP53 allelic state is critical in disease monitoring and represents an important prognostic stratification factor in MDS. Thus, TP53 allelic state characterization and the mutational burden evaluation must be considered as part of the MDS diagnostic workflow.
The potential consequence in our clinical case 3 is to consider allo-HCT, although TP53 state also translates to a dismal outcome with a higher relapse rate.43,49,50 Therefore, novel alternative therapeutic strategies such as APR-246, a p53 reactivator, are needed and currently under clinical investigation (Table 6). Preliminary results of a previous study in HMA-naïve patients with TP53 mutations and HR-MDS and AML showed that the combination of APR-246 plus azacitidine has promising clinical efficacy (complete remission rate of 56%) with deep molecular remission in all patients with a complete response.51
Is every high-risk patient the same?
The principal aims of treatment in HR-MDS are modifying the natural course of disease, limiting disease progression, and improving outcome.2 Initial stratification should primarily involve assessment of whether a patient is eligible for an intensified therapeutic approach including allo-HCT (Table 6). In patients with HR-MDS who have only mild, asymptomatic cytopenia, the treatment decision may be delayed in the absence of poor-risk mutations, and an intensified surveillance strategy could be considered.
Although the IPSS/R system was developed mainly to determine the prognostic risk in patients with newly diagnosed MDS, its value in predicting post-transplant outcome has been confirmed in several studies.52 Although already implemented in the existing scoring systems, karyotype abnormalities alone represent a significant risk factor for relapse, as do certain molecular abnormalities.43 Although not every complex karyotype is associated with an extremely poor prognosis, the presence of a TP53 mutation adds significantly to the adverse prognosis of these patients with HR-MDS. Thus, routine testing of TP53 and possible AML-defining mutations in patients with high-risk disease and complex karyotypes should be implemented.53
Interestingly, retrospective data in patients with HR-MDS and patients with AML treated with a 10-day decitabine regimen suggest that patients with TP53 mutations had higher response rates than patients with other mutations.54 Although data stating that TET2 mutational status may be a predictor of HMA treatment response exist, higher response rates did not translate into a survival benefit for these patients, and confirming prospective data are missing.55
A very smart and elegant new method will be the application of targeted and personalized drugs against specific molecular structures in MDS; here, the presence of druggable targets (Table 6) such as TP53 or IDH1/2 can be predictive of treatment response. However, targeted therapies such as APR-246, IDH1/2, or spliceosome inhibitors are just beginning to emerge. In fact, these targets may pave the way for holistic approaches covering the entire spectrum of myeloid neoplasms (Table 6).53
Integration of the “immunome” into personal risk stratification: a possible breakthrough?
In MDS, several mutations affecting the epigenetic modifiers (eg, TET2) or RNA splicing factors (eg, U2AF1) have been linked to NLRP3 inflammasome activation and enhanced innate immune signaling.56 Inflammatory cytokines are increased in the serum and bone marrow of these patients, and high cytokine levels correlate with a worse prognostic outcome.57 Consequently, our patient in clinical case 3 not only is exhibiting somatic mutations with higher risk for early disease progression (eg, TP53) but also displays mutations with a known high immunogenic impact (TET2, DNMT3A).
Inflammation within the bone marrow microenvironment in association with aberrant cellular immune responses evolving during MDS disease progression has independent prognostic value.58,59 Consequently, in addition to the incorporation of molecular data into current prognostic scoring systems, the inclusion of specific immunological data (“immunome”) could possibly further refine risk stratification in the future.58 Thus, a new immunologically based patient stratification model aiming to identify patients with prominent “autoinflammatory” features could be the next important step toward the development of personalized risk prediction models in MDS.58 A comprehensive clarification of the dysregulated immune pathways will enable patient stratification and, as a consequence, the development of new targeted drugs58 (Table 6).
Conclusion and future directions
Overall, increasing evidence exists that current MDS prognostic scoring systems are only an approximation in personal risk stratification. Thus, putting the puzzle together will require the integration of complex molecular and immunological interactions with clinical variables with the aim of establishing optimal personalized risk stratification models (Figure 1). Nevertheless, this also requires further expansion of our therapeutic armamentarium.
Correspondence
Uwe Platzbecker, Department of Hematology, Cellular Therapy and Hemostaseology, Leipzig University Hospital, Liebigstrasse 22, 04103 Leipzig, Germany; e-mail: uwe.platzbecker@medizin.uni-leipzig.de.
