
Abstract
Risk classification and tailoring of treatment are essential for improving outcome for patients with acute myeloid leukemia or high-risk myelodysplastic syndrome. Both patient and leukemia-specific characteristics assessed using morphology, cytogenetics, molecular biology, and multicolor flow cytometry are relevant at diagnosis and during induction, consolidation, and maintenance phases of the treatment. In particular, minimal residual disease (MRD) during therapy has potential as a prognostic factor of outcome, determination of response to therapy, and direction of targeted therapy. MRD can be determined by cell surface markers using multicolor flow cytometry, whereas leukemia-specific translocations and mutations are measured using polymerase chain reaction–based techniques and recently using next-generation sequencing. All these methods of MRD detection have their (dis)advantages, and all need to be standardized, prospectively validated, and improved to be used for uniform clinical decision making and a potential surrogate end point for clinical trials testing novel treatment strategies. Important issues to be solved are time point of MRD measurement and threshold for MRD positivity. MRD is used for stem cell transplantation (SCT) selection in the large subgroup of patients with an intermediate risk profile. Patients who are MRD positive will benefit from allo-SCT. However, MRD-negative patients have a better chance of survival after SCT. Therefore, it is debated whether MRD-positive patients should be extensively treated to become MRD negative before SCT. Either way, accurate monitoring of potential residual or upcoming disease is mandatory. Tailoring therapy according to MRD monitoring may be the most successful way to provide appropriate specifically targeted, personalized treatment.
Learning Objectives
Understand the current state of MRD as a tool to aid risk classification and tailor therapy accordingly
Perceive refinement of possibilities to improve stem cell transplantation outcome by MRD assessment
Case 1
A 52-year-old female without comorbidity presented with acute myeloid leukemia (AML). The karyotype was normal, and molecular testing revealed mutations in FLT3 (internal tandem duplication [ITD]; allelic ratio, 1.0), NPM1, and IDH2. She was enrolled into the National Cancer Research Institute AML19 study and received induction chemotherapy with daunorubicin, cytarabine, and 2 doses of gemtuzumab. After second induction, NPM1 mutant transcripts were detected (Figure 1A) in the blood and bone marrow aspirate, and the patient was allocated to intensification with CPX-351 (Vyxeos). On regeneration, transcript levels had increased by ∼1 log, and the patient was withdrawn from the trial protocol and received salvage therapy with the fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin regimen. On regeneration, a minimal residual disease (MRD) assessment showed a further ∼2-log increase in transcript levels, and the patient developed painless enlarged lymph nodes in the left side of the neck. A fine needle aspirate confirmed extramedullary disease. Quizartinib was initiated at a dose of 20 mg daily and was increased to 60 mg daily over a period of 3 weeks. A positron emission tomography–computed tomography scan confirmed that the extramedullary disease was confined to the left side of the neck, and the patient received 5 fractions of radiotherapy to sites of extramedullary disease and was scheduled for allograft from a matched sibling. Pretransplant MRD assessment showed an ∼2-log reduction in disease-related transcripts, and the patient proceeded to transplant. Conditioning was fludarabine (150 mg/m2 IV) and busulfan (12.8 mg/kg IV), and graft-versus-host disease (GvHD) prophylaxis was cyclosporin and mycophenolate. MRD assessment at posttransplant day +30 (D+30) was negative, weaning of immunosuppressive therapy was initiated, and quizartinib was restarted. Further MRD assessments at D+60 and D+100 were negative. The patient developed chronic GvHD affecting the skin and oral mucosa that was managed with extracorporeal photopheresis and topical steroids.
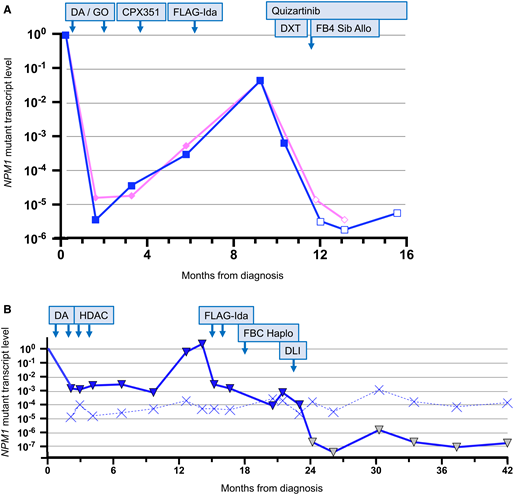
Examples of molecular MRD monitoring of NPM1 mutations before and after therapy. (A) Example of MRD-guided pretransplant management. See text for full case history. The pink line is peripheral blood and the blue line is bone marrow. The filled symbols indicate MRD positive at the level indicated. The empty symbols indicate MRD negative with the level of sensitivity indicated. (B) Example of MRD-guided therapy and peritransplant management. See text for full case history. The solid blue line is the bone marrow and the dotted blue line is the sensitivity. The filled blue symbols indicate MRD positive at the level indicated. The yellow symbols indicate MRD negative with the level of sensitivity indicated. DA, daunorubicin and cytarabine; DLI, donor lymphocyte infusion; DXT, radiotherapy; FBC Haplo, fludarabine and busulfan–conditioned haploidentical allograft and posttransplant cyclophosphamide; FB4 Sib Allo, fludarabine and busulfan–conditioned sibling allograft; FLAG-Ida, fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin; GO, gemtuzumab ozogamicin; HDAC, high-dose cytarabine.
Examples of molecular MRD monitoring of NPM1 mutations before and after therapy. (A) Example of MRD-guided pretransplant management. See text for full case history. The pink line is peripheral blood and the blue line is bone marrow. The filled symbols indicate MRD positive at the level indicated. The empty symbols indicate MRD negative with the level of sensitivity indicated. (B) Example of MRD-guided therapy and peritransplant management. See text for full case history. The solid blue line is the bone marrow and the dotted blue line is the sensitivity. The filled blue symbols indicate MRD positive at the level indicated. The yellow symbols indicate MRD negative with the level of sensitivity indicated. DA, daunorubicin and cytarabine; DLI, donor lymphocyte infusion; DXT, radiotherapy; FBC Haplo, fludarabine and busulfan–conditioned haploidentical allograft and posttransplant cyclophosphamide; FB4 Sib Allo, fludarabine and busulfan–conditioned sibling allograft; FLAG-Ida, fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin; GO, gemtuzumab ozogamicin; HDAC, high-dose cytarabine.
Case 2
A 23-year-old male with severe obesity presented with AML. The karyotype was normal, and molecular testing revealed a mutation in NPM1 only. He did not enter a clinical trial and received induction therapy with daunorubicin and cytarabine; after second induction, the bone marrow remained positive for disease-related transcripts; peripheral blood was not assessed. He received consolidation treatment with 2 cycles of high-dose cytarabine. MRD assessments performed on regeneration and at 3 and 6 months after therapy showed persistence of NPM1-mutant transcripts, and at 9 months there was a >2-log increase in expression levels (Figure 1B). This was confirmed on a second sample, and molecular progression was diagnosed. Salvage therapy with fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin was initiated, and after the second cycle there had been a 2- to 3-log reduction in the level of MRD, which remained positive. The patient proceeded with a haploidentical transplant with a parental donor; conditioning was fludarabine (160 mg/m2) and busulfan (12.8 mg/kg), and GvHD prophylaxis was cyclophosphamide (50 mg/kg on D+3 and D+5), tacrolimus, and mycophenolate. By D+100, all immunosuppression had been stopped, and an MRD assessment showed persistent NPM1-mutant transcripts. This was confirmed on a second sample, and persistent molecular disease was diagnosed. Donor lymphocyte infusion (DLI), at a dose of 105 cells per kilogram, was administered. After 4 weeks, the level of NPM1-mutated transcripts had decreased by 1 log, and after 8 weeks the patient tested negative for MRD. Molecular complete remission was maintained on several follow-up assessments up to 2 years posttransplant.
Introduction
With the improvement of therapeutic strategies in hematological malignancies, traditional morphological response definitions, such as complete remission,1 are increasingly insufficient because they do not take into account persistent malignant cells that are below the resolution of conventional techniques. Accurate measurement of this minimal or measurable residual disease is crucial for a more accurate prediction of relapse risk, which, in turn, can be used to inform treatment intensity. Most of the methods for MRD detection are broadly similar across the hematological malignancies (Table 1). The role of MRD in acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and multiple myeloma has been summarized elsewhere.2-6 In some ways, the assessment of MRD in AML is more challenging because of molecular and phenotypic heterogeneity, and MRD assessment is not yet completely standardized. This review focuses on the current status of MRD in AML and, in particular, the use of MRD for selecting patients for stem cell transplant and for pre- and posttransplant interventions.
Characteristics of the currently used MRD methods
| MRD method | Technique | Markers | Patient material | Leukemia type | Sensitivity | Advantage | Disadvantage | Relevant references |
|---|---|---|---|---|---|---|---|---|
| Molecular | ||||||||
| rearrangements | RT-qPCR | IGH | DNA | ALL | 10−4 to 10−5 | Patient specific | Requires diagnosis sample. Clonal evolution missed. | 4,5,64,65 |
| ddPCR | TCR | Myeloma | Standardized method | |||||
| CLL | ||||||||
| NGS | DNA | ALL | 10−3 to 10−6 | Does not require construction of patient-specific reagents; may detect clone shifts. | Expensive | 66 | ||
| Including clonoSEQ assay | Myeloma | |||||||
| CLL | ||||||||
| Fusion genes | RT-qPCR | BCR/ABL | RNA | ALL, CML | 10−4 to 10−5 | Standardized primers | Applicable in a limited number of patients | 32,33,56,67 |
| AML/ETO | AML | |||||||
| CBFB-MYH11 | AML | |||||||
| PML/RARa | APL | |||||||
| RUNX1/RUNX1T1 | ALL, AML | |||||||
| BCL3 translocations | CLL | |||||||
| Less common fusions | ||||||||
| Mutations | RT-qPCR | NPM1 | RNA | AML | 10−4 to 10−7 | Extremely sensitive | Applicable in a limited number of patients (∼30%). Clonal evolution missed. | 31,68 |
| NGS | Mutation panels | DNA | AML | 10−3 to 10−5 | Applicable in many patients | Requires further development for assay harmonization/standardization. Insensitive without error correction. | 37 | |
| May detect clone shift | ||||||||
| Overexpression | RT-qPCR | WT1 | RNA | AML | 10−5 | Sensitive | Applicable in a limited number of patients | 36,51,69,70 |
| Immunophenotypic | ||||||||
| Leukemia/myeloma associated immune phenotype | Distinctive antibody panels | Diagnosis and follow-up cells | AML | 10−4 | More informative with diagnosis sample | Clonal evolution missed. Immunophenotypic profiles required. | 71-73 | |
| ALL | Almost all patients | |||||||
| CLL | Relatively fast | |||||||
| Myeloma | ||||||||
| Different from normal | Distinctive antibody panels | Follow-up cells only | AML | 10−4 | Almost all patients. Relatively fast. | Extensive knowledge of normal and regenerating bone marrow required | 74 | |
| Leukemia stem cells | CD34+/CD38 including distinctive antibody panels | Diagnosis and follow-up cells | AML | 10−6 | High prognostic relevance to identify a poor risk group | Only CD34+ leukemia. Large number of cells required. | 61 | |
| MRD method | Technique | Markers | Patient material | Leukemia type | Sensitivity | Advantage | Disadvantage | Relevant references |
|---|---|---|---|---|---|---|---|---|
| Molecular | ||||||||
| rearrangements | RT-qPCR | IGH | DNA | ALL | 10−4 to 10−5 | Patient specific | Requires diagnosis sample. Clonal evolution missed. | 4,5,64,65 |
| ddPCR | TCR | Myeloma | Standardized method | |||||
| CLL | ||||||||
| NGS | DNA | ALL | 10−3 to 10−6 | Does not require construction of patient-specific reagents; may detect clone shifts. | Expensive | 66 | ||
| Including clonoSEQ assay | Myeloma | |||||||
| CLL | ||||||||
| Fusion genes | RT-qPCR | BCR/ABL | RNA | ALL, CML | 10−4 to 10−5 | Standardized primers | Applicable in a limited number of patients | 32,33,56,67 |
| AML/ETO | AML | |||||||
| CBFB-MYH11 | AML | |||||||
| PML/RARa | APL | |||||||
| RUNX1/RUNX1T1 | ALL, AML | |||||||
| BCL3 translocations | CLL | |||||||
| Less common fusions | ||||||||
| Mutations | RT-qPCR | NPM1 | RNA | AML | 10−4 to 10−7 | Extremely sensitive | Applicable in a limited number of patients (∼30%). Clonal evolution missed. | 31,68 |
| NGS | Mutation panels | DNA | AML | 10−3 to 10−5 | Applicable in many patients | Requires further development for assay harmonization/standardization. Insensitive without error correction. | 37 | |
| May detect clone shift | ||||||||
| Overexpression | RT-qPCR | WT1 | RNA | AML | 10−5 | Sensitive | Applicable in a limited number of patients | 36,51,69,70 |
| Immunophenotypic | ||||||||
| Leukemia/myeloma associated immune phenotype | Distinctive antibody panels | Diagnosis and follow-up cells | AML | 10−4 | More informative with diagnosis sample | Clonal evolution missed. Immunophenotypic profiles required. | 71-73 | |
| ALL | Almost all patients | |||||||
| CLL | Relatively fast | |||||||
| Myeloma | ||||||||
| Different from normal | Distinctive antibody panels | Follow-up cells only | AML | 10−4 | Almost all patients. Relatively fast. | Extensive knowledge of normal and regenerating bone marrow required | 74 | |
| Leukemia stem cells | CD34+/CD38 including distinctive antibody panels | Diagnosis and follow-up cells | AML | 10−6 | High prognostic relevance to identify a poor risk group | Only CD34+ leukemia. Large number of cells required. | 61 | |
CML, chronic myeloid leukemia; ddPCR, droplet digital PCR; NGS, next-generation sequencing; RT-qPCR, reverse-transcription quantitative polymerase chain reaction.
Risk classification at diagnosis
Detailed characterization of patients using a range of diagnostic techniques is essential for optimal treatment of AML.7 Morphology remains the cornerstone of diagnosis and can distinguish different subtypes based on cellular and differentiation features,8 most obviously the M3 FAB subtype, which allows initiation of urgent therapy, including all-trans retinoic acid.9 Flow cytometry is mandatory to confirm the diagnosis of AML, and it can be used to assign subtype based on specific cell surface markers that are expressed in particular phases during the differentiation of hematopoietic cells (ie, CD markers).8 Although there is no clear relationship between immunophenotype and outcome, multicolor flow cytometry (MFC) allows rapid determination of cell surface antigen expression status, which is critical given the increasing availability of immunotherapies (eg, gemtuzumab ozogamicin). MFC can also provide early clues to the underlying cytogenetic and molecular lesion (eg, APL and NPM1-mutated AML have characteristic immunophenotypes).10,11 Cytogenetic analysis provides the most powerful prognostic information in AML and, a full karyotype, complemented by fluorescence in situ hybridization, is needed to identify recurrent chromosome abnormalities that are strongly predictive of outcome and inform the European LeukemiaNet (ELN) guidelines for risk classification.7 Molecular genetic analysis provides further essential prognostic information12 that is particularly informative in patients with a normal karyotype (eg, the patients described here). In addition to risk group assignment, molecular analysis is increasingly important for selection of patients for targeted therapies, including small molecule inhibitors of FLT3, IDH1, and IDH2, which are becoming widely available.
In most AML clinical studies, the majority of patients are classified as intermediate risk; despite detailed molecular analysis at diagnosis, outcome prediction remains imperfect. Therefore, particularly in this group, treatment-emergent factors, such as MRD, may be particularly informative with respect to the selection of appropriate consolidation therapy.13
MRD assessment during treatment
The majority of AML patients treated with induction chemotherapy achieve a morphological complete remission (ie, <5% blasts by morphology1 ); however, this is not a very sensitive method to accurately determine the residual load of leukemia cells.13,14 More sophisticated methods for detection of residual disease can provide sensitivity that is orders of magnitude greater than that achieved by morphology. MRD status during treatment effectively provides a read-out of multiple patient- and leukemia-specific characteristics, not all of which are well understood. There are several methods to detect MRD in blood and bone marrow of AML patients during therapy; these include MFC, amplification of leukemia-specific transcripts by reverse-transcription quantitative polymerase chain reaction (RT-qPCR)15-20 and, more recently, detection of leukemia-specific mutations using next-generation sequencing (NGS).21-23 Details about the applicability of the different methods are summarized in Table 1.
MRD status has been robustly correlated with risk for relapse in many independent large clinical trials. The prognostic value has been shown as early as after the first cycle of treatment. Many studies use MRD status after the second cycle of treatment to further refine risk classification, because this time point has significant prognostic value and still allows enough time to initiate logistics for stem cell transplantation (SCT) when necessary.
Flow cytometric MRD can assess the efficacy of induction/consolidation or salvage on the dominant diagnostic (or relapse) leukemic blast populations by identifying leukemia associated immune phenotypes (LAIPs) on ≥10% to 20% of leukemic blasts in the diagnostic (or relapse) sample and monitoring these specific LAIPs during therapy.24 Because the LAIP is based on aberrant expression of CD markers on the cell surface that are not present in healthy bone marrow, residual leukemia cells can also potentially be detected during therapy without knowledge of the diagnostic (or relapse) LAIPs. This is referred to as the “different from normal” (DfN) approach, and it can be applied when no diagnostic (or relapse) samples are available (eg, when patients are referred to a transplant center without having been monitored there previously). Additionally, the DfN approach can detect any phenotypically aberrant leukemia subpopulations that may have been minor or undetectable at diagnosis but have expanded during therapy, potentially due to clonal evolution, such as that observed following transplants.25,26 Therefore, the recommendation of the ELN is to combine flow cytometric MRD methods instead of limiting CD marker panels to the LAIPs found at diagnosis.27 In the recently completed HOVON 132 trial, all markers were measured so that when clinical data become available, the potential relevance of novel upcoming clones can be established. However, the DfN approach can lead to false-positive results and, therefore, reduced specificity, particularly when there is insufficient knowledge of progenitor phenotypes resulting from regeneration of the bone marrow after chemotherapy or transplant. Tracking the diagnostic or relapse LAIPs as biomarkers of the major pretreatment leukemic populations may also be more appropriate when assessing the efficacy of novel treatment strategies to reduce the dominant leukemic clones. At present, however, flow cytometric MRD assays are not sufficiently standardized and validated to be used as a surrogate end point. Joint efforts of current MRD assessments implemented in the majority of clinical trials should facilitate this. Several groups are currently collaborating to standardize (where needed) and harmonize (where possible) the flow cytometric MRD methods.28 Additionally, several studies are currently directed toward computational approaches to objectify, simplify, and speed up MRD assessments.29
Molecular MRD analysis provides a highly sensitive alternative to MFC in patients with a validated target for RT-qPCR (ie, patients with recurrent in-frame gene fusions or NPM1 mutations, together accounting for ∼60% of younger adults).30 For patients with NPM1-mutated AML, molecular MRD analysis has demonstrated remarkable discrimination in a number of large prospective clinical trials and is the most powerful prognostic factor for these patients,16,17,31 identifying those who will benefit from upfront transplantation. Apart from NPM1, prognostic impact of PML-RARA,32 RUNX1-RUNX1T1, and CBFB-MYH1115,19 fusion transcripts during treatment and follow-up is well established, and the relevance of transcript status for other rarer fusion genes is currently being investigated in large prospective trials, such as NCRI AML19 and MyeChild01. Levels of expression of these leukemia-specific transcripts at diagnosis vary markedly, and this impacts on assay sensitivity33 (eg, NPM1-mutant transcripts are highly expressed at diagnosis, resulting in sensitivity of up to 1:107, whereas assays to monitor KMT2A fusions may only afford sensitivity orders of magnitude lower).34 Molecular markers considered unsuitable on their own to monitor MRD include FLT3, because of its relative instability at relapse.35
Assays for WT1 mutation and expression are generally no longer considered satisfactory for MRD measurement; although often upregulated or mutated at relapse, expression is insufficiently specific, whereas mutation status is insufficiently sensitive, to reliably detect relapse.36
The most valuable addition to the current battery of assays33 would be the ability to measure all AML-associated mutations with the sensitivity required for MRD analysis using NGS. Therefore, many initiatives are currently ongoing to improve sensitivity and reproducibility of NGS MRD; these will need to be completed before NGS can be uniformly introduced for MRD-directed treatment allocation for clinical trials and in routine practice.37 In addition to harmonization of the MRD assays, the combination of MFC and molecular analyses needs further evaluation, because it has recently been shown that these methods are complementary.23
All current AML MRD platforms would benefit from further standardization and from unified criteria for MRD positivity. Considering the progress in this qualification effort,38 it is anticipated that MRD might be accepted by the U.S. Food and Drug Administration as a surrogate end point for treatment response in the near future.
In addition to improvements in the methods for MRD detection, uncertainties remain regarding thresholds for MRD positivity across AML subtypes, treatment strategies, and informative time points, as well as the utility of MRD by monitoring in peripheral blood samples.39-41 These are currently still under investigation and should become apparent within the next few years.
Use of MRD to inform pretransplant management
Using a combination of comprehensive diagnostic profiling and MRD status, patients who are likely to be cured with chemotherapy alone and who can be safely monitored in first complete remission can be discriminated from those who will benefit from upfront transplantation.16,31 Although there are no randomized data to support this approach, based on currently available nonrandomized data, this approach has generally been adopted and is implemented in a number of large clinical trials (Table 2). Ongoing prospective studies will provide further information to guide decision making; in particular, the UK NCRI “monitor vs no monitor” randomization will report next year and will be informative regarding the benefit of MRD-directed consolidation therapy.
Clinical validation of MRD before and after transplant
| Study | Patients (N) | Patient group | MRD method | Time point | Threshold | Outcome | References |
|---|---|---|---|---|---|---|---|
| Meta-analysis | 1431 | Adult non-APL | MFC and molecular | Pretransplant | 0.1% for FCM | Prognostic for relapse and survival | 45 |
| EORTC/GIMEMA | 81 | MRD+ eligible for SCT | MFC | After consolidation | 0.035% | No contraindication for allo-SCT in MRD+ patients | 75 |
| IRCCS Genoa | 224 | Transplanted in CR1/CR2 | MFC and WT1 | Pretransplant | 2.5 × 10−4 250 copies/Abl × 104 | Prognostic value of MRD for pre- and posttransplant interventions | 69 |
| Toronto/Korea | 104 | Adult AML eligible for transplant | NGS | Diagnosis and pre- and posttransplant (day 21) | VAF0.2%-post-HCTD21 | Good prediction of relapse | 76 |
| Seattle | 279 | Adult | MFC | Pre- and posttransplant | 0.1% | MRD+ poor prognosis irrespective of myeloablative conditioning | 77 |
| Hannover | 116 | >18 y | NGS | In CR before transplant | Error corrected | Predictive for relapse and survival Refine SCT | 21 |
| RELAZA2 | 60 | MRD+ >18 y | NPM1 fusion gene, CD34 Chimerism MFC | After induction/consolidation Preemptive | Depending on test used | Predictive for relapse | 50 |
| Study | Patients (N) | Patient group | MRD method | Time point | Threshold | Outcome | References |
|---|---|---|---|---|---|---|---|
| Meta-analysis | 1431 | Adult non-APL | MFC and molecular | Pretransplant | 0.1% for FCM | Prognostic for relapse and survival | 45 |
| EORTC/GIMEMA | 81 | MRD+ eligible for SCT | MFC | After consolidation | 0.035% | No contraindication for allo-SCT in MRD+ patients | 75 |
| IRCCS Genoa | 224 | Transplanted in CR1/CR2 | MFC and WT1 | Pretransplant | 2.5 × 10−4 250 copies/Abl × 104 | Prognostic value of MRD for pre- and posttransplant interventions | 69 |
| Toronto/Korea | 104 | Adult AML eligible for transplant | NGS | Diagnosis and pre- and posttransplant (day 21) | VAF0.2%-post-HCTD21 | Good prediction of relapse | 76 |
| Seattle | 279 | Adult | MFC | Pre- and posttransplant | 0.1% | MRD+ poor prognosis irrespective of myeloablative conditioning | 77 |
| Hannover | 116 | >18 y | NGS | In CR before transplant | Error corrected | Predictive for relapse and survival Refine SCT | 21 |
| RELAZA2 | 60 | MRD+ >18 y | NPM1 fusion gene, CD34 Chimerism MFC | After induction/consolidation Preemptive | Depending on test used | Predictive for relapse | 50 |
ABL, Abelson gene; allo-SCT, allogeneic SCT; CR, complete remission; FCM, flow cytometry post-HCTD21, day 21 after hematopietic cell transplantation; VAF, variant allele frequency.
Thus, MRD positivity is a marker for selection of patients for allogeneic SCT. However, it has been shown that the risk of posttransplant relapse is lowest in MRD-negative patients21,42-46 ; therefore, the question is whether MRD-positive patients would benefit from intensification of treatment before SCT to convert to MRD negativity. Although superficially attractive, the potentially drug-resistant MRD-positive bone marrow may not easily become MRD negative, and this could lead to additional toxicity without benefit, which could potentially compromise long-term survival. There are currently no prospective data to inform such decisions; however, ongoing studies, such as UK NCRI COSI, may be informative in this regard. In the absence of such data, individual patient management can be informed by sequential MRD measurement, and this is potentially an opportunity for the use of novel relatively nontoxic targeted agents for the elimination of drug-resistant cell reservoirs prior to transplant.
It also remains unclear whether augmented conditioning can improve the outcome for patients who are MRD positive before transplant; retrospective studies comparing myeloablative and reduced-intensity conditioning for these patients have yielded conflicting results.46,47 A retrospective study has suggested an advantage for MRD-positive patients who receive umbilical cord blood transplants,48 although this remains controversial49 ; importantly, this approach removes the option of using DLI after transplant. Prospective studies to define the optimal management for these patients are urgently required.
After SCT
For patients who remain persistently MRD positive after transplant, relapse is inevitable without further intervention. Treatment options depend on the clinical situation and time from transplant. Data to inform management of MRD positivity in the posttransplant setting are fairly sparse, and there is only 1 published prospective study to date. The RELAZA2 study demonstrated that azacitidine can prevent or delay hematological relapse in a proportion of patients with ongoing MRD positivity after treatment, and this may be more effective in patients who have been transplanted, potentially indicating an immunological effect of this treatment.50 Posttransplant MRD status may also be useful to plan withdrawal of immunosuppression; after this has been tapered, persistent MRD positivity should prompt consideration of DLI. A number of studies have reported very good success rates in this setting.51,52 It appears that DLI can convert patients to MRD negativity, and long-term disease-free survival rates of 80% to 100% have been reported in these small retrospective studies. Certainly, the success rates for the use of DLI in the MRD setting appear to be significantly higher than when DLI is used at hematological relapse.53,54 Azacitidine, in combination with DLI, has also been reported to be effective in this situation54 and could be considered if DLI alone fails or the level of MRD is very high. DLI is also reported to have activity in posttransplant MRD positivity in other hematological malignancies, such as ALL55 and CML.56 Based on these effective options for persistent or re-emergent MRD positivity, serial MRD monitoring after transplant is recommended,57 particularly for patients with a sensitive molecular marker for whom impending relapse can be predicted months in advance, providing a time window for intervention.34 We suggest that posttransplant surveillance is continued for ≥1 to 2 years after transplant because this period is associated with the highest risk of relapse. A proportion of patients will be unable to receive DLI because of the donor status or the presence of GvHD, and some of these patients will not respond to or will be unable to tolerate azacitidine. For these patients, management is more challenging. In the absence of evidence, novel agents could be considered (eg, FLT3 or BCL2 inhibitors). Further prospective clinical trials that use MRD-directed posttransplant interventions are now required. Some studies that are in progress should give further insights in the near future; in this respect, HOVON recently initiated a prospective phase 3 trial to determine the efficacy of panobinostat maintenance therapy vs standard of care after allogeneic SCT, which includes MRD assessment before and at several time points after SCT.
Case discussions
Case 1
This patient was ELN intermediate risk based on molecular and cytogenetic features at diagnosis; however, failure to achieve MRD negativity in the peripheral blood after second induction is associated with a very high risk of relapse.20 For patients with FLT3 ITD, pretransplant MRD positivity is associated with a high risk for posttransplant relapse58 ; therefore, intensification was attempted but was unsuccessful and resulted in molecular and extramedullary progression indicating chemorefractory disease. In this situation, novel targeted agents may be useful to reduce disease burden prior to, and to sustain remission after, transplantation, as illustrated by the effect of quizartinib in this case.
Case 2
This patient was ELN favorable risk based on molecular and cytogenetic features at presentation; however, at the end of treatment he remained persistently MRD positive, indicating impending relapse. Due to a high risk for treatment-related mortality, intervention was only undertaken after a significant increase in MRD levels. Although molecular complete remission was not achieved after salvage therapy, patients who are FLT3 ITD negative with low levels of NPM1-mutant transcripts have a generally good outcome after transplant.58 Despite this, he remained MRD positive after withdrawal of immunosuppression. DLI resulted in a rapid clearance of residual disease, providing an example of the graft-versus-leukemia effect for eliminating MRD after transplant.
Future perspectives
Risk classification based on diagnostic cytogenetic and molecular characterization and refined by MRD assessment at early time points is critical for proper clinical decision making in AML. MRD provides the most powerful predictor of outcome in intermediate risk AML, and its measurement in clinical trials and everyday practice is strongly recommended.57 MRD data can be exploited to tailor treatment intensity according to response and, as shown here, they can serve as a trigger for application of novel therapies (eg, FLT3 inhibitors, IDH1/2 inhibitors, splicing modulators, or epigenetic modifiers).38,59
Although current MRD platforms provide very powerful prognostic information, further improvement is possible through standardization of assays and accumulation of larger data sets. These collaborative efforts will lead to a clearer definition of the optimum time points, sample sources, and thresholds for clinical decision making. With regard to MFC MRD, a deeper understanding of the characteristics of relapse-initiating cells may further improve prognostic value.60,61 A recently designed 1-tube assay to assess leukemia stem cell load is used in HOVON studies, which takes clonal evolution into account and which is associated with clinical outcome at diagnosis and during therapy.62 In addition, it has been shown that combining molecular and MFC data also aids in distinguishing a very poor risk group who may benefit from intensified treatment strategies.61
It is currently being investigated whether MRD can be used as surrogate end point in clinical trials to assess efficacy of treatment. This might considerably improve the development of new treatment options for the patient subgroups who are most likely to benefit from the intervention.63
Finally, although few studies have been published regarding the use of MRD results to optimize peritransplant management, this now appears to be an extremely promising field of study with the potential to test MRD-triggered interventions that could have a major impact on the rate of posttransplant relapse by focusing interventions on those patients at highest risk. Results of these emerging studies are eagerly anticipated.
Acknowledgments
The authors thank their colleagues from the Dutch MRD working group and from ELN for their sustained collaboration on this work, as well as S. Freeman for valuable input into the manuscript.
The research at Amsterdam UMC was supported by the Dutch Cancer Society (G.O.).
Correspondence
Jacqueline Cloos, Amsterdam UMC, Location VUMC, Cancer Center Amsterdam, de Boeleaan 1117, 1081 HV, Amsterdam, The Netherlands; e-mail: jcloos@amsterdamumc.nl.
