
Abstract
Despite the considerable improvements witnessed over the last few decades in the feasibility and safety of allogeneic hematopoietic cell transplantation (allo-HCT) for hematological malignancies, disease relapse continues to represent a frequent occurrence, with largely unsatisfactory salvage options. Recent studies have shed new light on the biology of posttransplantation relapses, demonstrating that they can frequently be explained using an evolutionary perspective: The changes in disease clonal structure and immunogenicity that are often documented at relapse may in fact represent the end results of a process of selection, allowing the outgrowth of variants that are more capable of resisting the therapeutic control of allo-HCT. This review provides an overview of the mechanisms forming the basis of relapse, including clonal evolution, gain of tropism for privileged sites, genomic and nongenomic changes in the HLA asset, and enforcement of immune checkpoints. Finally, this review discusses how these mechanisms may combine in complex patterns and how understanding and untangling these interactions may provide key knowledge for the selection of personalized therapeutic approaches.
Learning Objectives
Understand the biology governing posttransplantation relapse and the relative contribution of oncogenic and immune-related drivers
Translate knowledge of relapse biology into rationales for the selection of available therapies and for the development of new approaches
Clinical case
A 43-year-old man was diagnosed with de novo acute myeloid leukemia (AML). Disease characterization at the time of diagnosis documented trisomy of chromosome 21 as the sole cytogenetic abnormality, a high allele ratio internal tandem duplication (ITD) of the FMS-like tyrosine kinase 3 (FLT3) gene, and a type A frameshift mutation of nucleophosmin 1 (NPM1 mutA). After standard induction chemotherapy, he achieved morphological remission with persistent positivity of NPM1 mutA, and after 2 cycles of consolidation, the mutation remains positive, although at very low titer.
The patient then received an allogeneic hematopoietic cell transplantation (allo-HCT) from his HLA-haploidentical brother with use of myeloablative conditioning, peripheral blood stem cells as the graft source, and posttransplant cyclophosphamide followed by tacrolimus plus mycophenolate mofetil as graft-versus-host disease (GVHD) prophylaxis.
He did not experience acute GVHD, and given the molecular persistence of NPM1 mutA at the bone marrow evaluation on day 30 after allo-HCT, immunosuppression was rapidly tapered and fully discontinued at day 90, when the bone marrow evaluation detected, for the first time, negativity for NPM1 mutA. In the following months, the patient remained in molecular remission and developed a limited nonsevere chronic GVHD, with complete regression after a short course of topical steroids. At 15 months after allo-HCT, owing to a consistent drop in platelet and white blood cell counts, a bone marrow evaluation was repeated, documenting disease relapse.
Generalities about relapse
Over the last few decades, the outcome of allo-HCT for hematological malignancies has considerably improved.1 This is mostly explained by a significant reduction in transplant-related mortality, achieved through the development of reduced-toxicity conditioning regimens, more effective anti-infectious drugs, and novel tools for the control of GVHD. Conversely, over the same time frame, the incidence of posttransplantation disease recurrence has remained largely unchanged. To make matters worse, the prognosis of patients who experience relapse also has not improved, with fewer than 20% of relapsing patients able to achieve long-term survival. As a consequence, in all published series, relapse currently represents the leading cause of mortality after allo-HCT.2
To date, the only practical measures to reduce relapse mortality are to increase the depth of pretransplant remission and to anticipate relapse treatment to the minimal residual disease stage. Still, this is not always possible, owing to the intrinsic chemoresistance of malignant stem cells and to the rapid growth kinetics of diseases such as acute leukemia, ultimately leaving patients with overt hematological recurrence in dire need of new options.
This review discusses how improved knowledge of the biology of hematological malignancies has provided new tools to investigate the mechanisms forming the basis of posttransplantation relapses, leading to appreciation that recurrences after allo-HCT are fundamentally different from their counterparts occurring after conventional chemotherapy, and guiding the development of more rational and personalized therapeutic approaches.
Posttransplantation clonal evolution
Although the concept of cancer clonal evolution was theorized several decades ago in a landmark perspective by Peter Nowell,3 it was only with the advent of next-generation sequencing (NGS) that it became possible to analyze this phenomenon in fine detail. Hematological malignancies often grant the possibility to easily collect tumor samples longitudinally during disease history, thus representing one of the best models to study clonal evolution, both under neutral conditions and under strong selective pressure, such as the one imposed by chemotherapies or targeted therapies.
A number of reports have convincingly shown that, upon intensive chemotherapy, the clonal structure of hematological malignancies can undergo dramatic modifications, changing the relative proportions of disease subclones through disappearance of major and minor subclones and independent acquisition of new genomic alterations in one or more subclones.4,5 Although in some instances the changes in clonal structure observed at relapse represent only the end result of the interaction between a genetically heterogeneous disease and a semirandom sweep eliminating the large majority of its initial bulk, in most cases the genetic features of the relapsed tumor are evolutionarily selected and can provide precious hints of mechanisms of resistance to therapy and, indirectly, of disease vulnerabilities.
Allo-HCT represents an intriguing setting in which to investigate clonal evolution because it combines 2 very different therapeutic mechanisms that can both impact evolutionary dynamics dramatically: the rapid debulking operated by the conditioning regimen and the long-lasting immune effects associated with the transfer of a partially incompatible immune system from the donor to the patient (graft-versus-tumor [GVT] effect) (Figure 1).6
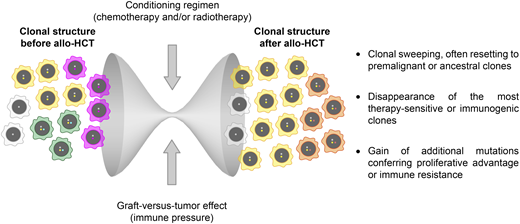
Determinants and consequences of clonal evolution after allogeneic hematopoietic cell transplantation.
Determinants and consequences of clonal evolution after allogeneic hematopoietic cell transplantation.
Initial studies, based on standard cytogenetics or single-nucleotide polymorphism arrays and focused mainly on patients with AML, documented the frequent appearance of new chromosomal deletions, duplications, or copy-neutral losses of heterozygosity (CN-LOH) in posttransplantation relapses.7-9
With the sole relevant exception of alterations encompassing the HLA region (discussed in detail in other sections of this review), most of the medium- to large-scale alterations detected in these studies occurred in regions that are recurrently mutated also at the time of diagnosis or at relapse after sole chemotherapy, suggesting a limited role of the immune effects of allo-HCT in driving their appearance. This does not mean, however, that such alterations do not carry clinically relevant implications. For instance, the CN-LOH of chromosome 13q that is frequently documented in posttransplantation AML relapses is accompanied in virtually all cases by doubling of the allelic burden of a preexisting FLT3-ITD, rendering the disease more aggressive but also more dependent on FLT3 signaling, and possibly more vulnerable to its therapeutic inhibition.8,9
More recent studies based on NGS have captured posttransplantation genomic changes in much finer detail, tracing the evolutionary trajectories also of point mutations and small indels. Thanks to this higher resolution, it is now even more evident not only that, in the majority of relapsing patients, tumor cells carry new mutations and/or have lost some of the ones that were present at diagnosis, but also that the overall landscape of these mutations is not significantly different between the 2 time points, with no evidence of mutations uniquely detected at posttransplantation relapse.10,11 It should be noted that the genetic architecture of the underlying disease appears to have a significant influence on the type of clonal evolution observed at relapse; in fact, it has been reported that malignancies with higher intratumoral heterogeneity (such as myelodysplastic syndromes) often relapse through the expansion of minor subclones,12 whereas genetically simpler cancers (such as myelofibrosis) conversely relapse through limited clonal evolution with an overall net loss of mutations.13
Extramedullary relapses
One of the very first hints suggesting that posttransplantation relapses might have a unique underlying biology was the observation that after allo-HCT a sizable proportion of patients experience recurrence exclusively in extramedullary (EM) sites.14-16 Whereas isolated EM disease represents an extremely rare initial presentation of both AML and acute lymphoblastic leukemia, in both malignancies it can account for up to 10% of posttransplantation relapses. The most common sites for isolated EM relapses are the skin and soft tissues for AML and the central nervous system for acute lymphoblastic leukemia. Although it is intuitive to associate isolated EM relapses with the looser immunosurveillance of “sanctuary” sites, mechanistic studies supporting this hypothesis are largely lacking, and most hints of their biology come from clinical retrospective studies.
It has been documented, for instance, that isolated EM relapses tend to occur later after allo-HCT than do bone marrow relapses, hinting that EM relapses might stem from a very small population of tumor cells, evolutionarily selected for their tissue tropism rather than for their proliferative fitness.14 In addition, most studies agree that the presence of EM disease at diagnosis is a strong risk factor for experiencing EM relapse, suggesting that the tropism for EM sites may at least in part be disease intrinsic. Finally, several reports documented a significant association between chronic graft-versus-host disease (cGVHD) and isolated EM relapses (although, interestingly, in some studies, cGVHD appeared protective, whereas in others, it represented a risk factor), but in the same analyses, the association with acute GVHD was not significant. This is also in line with data available for the association between GVHD and bone marrow relapses, suggesting that the duration of the exposure to T-cell alloreactivity may be more relevant than its intensity in shaping relapse dynamics. Regarding the possible explanation of the conflicting results for the association between cGVHD and isolated EM relapses, it should be considered that whereas cGVHD might indeed bring higher numbers of T cells in EM sites, in the long term, development of tissue fibrosis and prolonged use of systemic immunosuppressive therapies might actually result in an immunoprivileged environment favorable for the seeding and outgrowth of malignant cells.
Posttransplantation immune evasion
The establishment of a proficient GVT effect after allo-HCT depends on a complex network of interactions between the donor-derived immune system, residual tumor cells, and the host microenvironment, and there is growing evidence that alterations in each of these players may be involved in the occurrence of relapse.17 In particular, the interplay between donor-derived T cells and host malignant cells has been shown to be one of the cornerstones of the GVT effect. In addition to their physiological recognition of “foreign” tumor-associated antigens and cancer neoantigens, donor T cells can also respond against nonpathological patient-specific antigens, including minor histocompatibility antigens and mismatched HLA molecules, and many clinical and laboratory studies have convincingly demonstrated that T-cell alloreactivity against these non–self-antigens is possibly even more relevant in driving the GVT effect.18,19 In particular, incompatible HLA molecules are the most potent targets of primary T-cell alloreactivity, capable of eliciting responses several orders of magnitude greater than any peptidic antigen.20 Still, whereas the role of T-cell alloreactivity against incompatible HLA molecules in prompting GVHD goes unquestioned, its contribution to the GVT effect is more difficult to ascertain from clinical data and is mostly supported by experimental models or indirect evidence, such as the documentation of genomic loss of incompatible HLAs in leukemic cells at the time of relapse (commonly referred as HLA loss).
HLA loss
The molecular mechanism of HLA loss is in the majority of cases a large CN-LOH event encompassing the entire HLA complex and resulting in the permanent and irreversible loss of all incompatible class I and class II alleles. However, in this type of genomic rearrangement, the loss of one haplotype is rapidly counterbalanced by the compensatory duplication of the remaining (compatible) one; thus, genomic HLA loss changes the type, but not the quantity, of the HLA molecules expressed by leukemic cells (Figure 2, left column).2,21,22

As a result, donor T cells that after allo-HCT progressively become enriched in anti–patient HLA specificities, will then encounter on the surface of leukemic cells only “self” HLA alleles, catalyzing much weaker immune responses and allowing leukemia to outgrow in clinically evident relapse. This mechanism of leukemia immune evasion and posttransplantation relapse was first described in the context of allo-HCT from family haploidentical donors, where up to 30% of relapses are HLA loss,21,23-25 and has only more recently been documented also after allo-HCT from partially HLA-mismatched unrelated volunteer donors, where the incidence of relapses is reportedly lower (10% to 15%, based on the small series published to date).8,26 This difference in incidence appears proportional to the number of donor–recipient HLA incompatibilities in the 2 settings, suggesting that a higher number of mismatches will likely convey a stronger anti-HLA T-cell response and, as a consequence, prompt leukemia to evade recognition by using this strategy. Intriguingly, unrelated umbilical cord blood transplantation might represent an exception to this model. In fact, despite the multiple HLA mismatches commonly accepted for umbilical cord blood HCT, relapses in this setting do not seem to occur through HLA loss,27 fostering more in-depth studies on the immune interactions that characterize this highly unique HCT setting. Of interest, similarly to EM relapses, HLA loss also tends to be observed more frequently in late relapses,21,23 hinting at a long selection process starting from a very small clone.
Besides offering important indirect insights into the biological mechanisms of the GVT effect, documentation of HLA loss at posttransplantation leukemia relapse also has some relevant implications for clinical practice, and for this reason, it is highly recommended in the European Society for Blood and Marrow Transplantation guidelines for the management of relapsing AML.28 Detection of HLA loss at relapse was initially based on rather cumbersome techniques, including HLA typing and single-nucleotide polymorphism array analysis of purified leukemic cells,21,22,24,25 but it has recently been made easier by the development of HLA-KMR (GenDx, Utrecht, the Netherlands), a rapid and reliable quantitative polymerase chain reaction–based assay allowing the differential diagnosis of HLA loss also in nonpurified samples,29 as well as by the even more recent development of NGS-based technologies.27
The most immediate consequence of documenting HLA loss at posttransplantation relapse is that therapeutic approaches leveraging the original donor T cells, such as rapid tapering of immunosuppressants or performing donor lymphocyte infusions (DLIs), will possibly have very limited, if any, effect against the relapsed disease.23 Still, these therapeutic strategies will maintain an unchanged risk of inducing GVHD and thus must be avoided until HLA loss has not been ruled out. Conversely, when HLA loss is confirmed, salvage therapies should be aimed at circumventing this immune evasion mechanism, using strategies that either target the retained (compatible) HLA molecules or do not rely at all on conventional T cell–mediated target recognition. One example of the first concept is offering a second haploidentical HCT to patients with HLA loss, purposely choosing a donor who is mismatched for the HLA haplotype that has been duplicated by leukemic cells and thus is expectedly able to respond robustly against them.30 A recent study hinted the promising potential of this approach, fostering larger prospective confirmatory trials.31 Alternatively, a number of immunotherapeutic approaches that are currently under clinical development do not leverage conventional T-cell receptor (TCR)–HLA interactions and may find a specific rationale in patients with HLA loss relapses. Some examples of these strategies include adoptive immunotherapy with non–HLA-restricted immune cells (including natural killer cells, cytokine-induced killer cells, and CD1-restricted lymphocytes) or redirection of conventional T-cell specificity by the use of bispecific antibodies or chimeric antigen receptors.
Downregulation of HLA class II molecules
More recently, another modality of altering the repertoire of HLA molecules has been described in posttransplantation AML relapses. In this case, malignant cells downmodulate the expression on their surface of both compatible and incompatible HLA class II molecules (HLA-DR, -DQ, and -DP) while retaining normal levels of expression of HLA class I molecules (Figure 2, middle column).9,11,32 Noticeably, loss of expression of HLA class II molecules appears sufficient to abrogate immune recognition of leukemia, supporting previous studies in patients and animal models that suggested a central role of the interactions between CD4 T-cell and HLA class II molecules in the GVT effect.33,34
The mechanism at the basis of this quantitative alteration of HLA expression has yet to be elucidated, but available data point to a central involvement of class II major histocompatibility complex transactivator (CIITA), the master regulator of HLA class II expression. Notably, genetic analysis of these cases of relapse did not detect mutations in either CIITA or other regulators of antigen presentation, suggesting that the primary drivers of this mechanism of relapse might be epigenetic. Indeed, a number of reports have already evidenced multiple layers of epigenetic regulation of HLA class II expression, both through DNA methylation and through histone modifications,35 warranting new studies to investigate how these features change in hematological malignancies after allo-HCT.
Although data on this relapse modality are still limited, it would appear to have a prevalence similar to genomic HLA loss (30% to 40% of relapses), but, in contrast to the latter, it does not seem to show a significant correlation with the number of donor–recipient HLA incompatibilities, occurring with similar frequency in both matched and mismatched allo-HCTs.9,11 This partly surprising observation suggests that the downregulation of HLA class II molecules may serve malignant cells mostly to escape from responses directed toward HLA class II–restricted minor histocompatibility antigens (reportedly much more numerous than those presented by class I molecules) rather than to evade direct alloreactivity against incompatible HLAs.
From a clinical perspective, documentation of downregulated HLA class II (which can be performed easily with conventional flow cytometry) also has direct practical implications. Although, as in genomic HLA loss, donor-derived T cells are unable to directly recognize and eliminate these relapse variants, the nongenomic nature of this alteration implies that it may be reversible. In particular, it has been shown that exposure of relapsed leukemia to interferon-γ can activate an alternative promoter of CIITA and recover the surface expression of HLA class II on leukemic cells.9,11 Although it is currently not possible to directly administer this cytokine to relapsing patients, it is possible to induce its physiologic release by immune cells in response to other targets, and this may in turn recover expression of HLA class II on leukemic cells and its consequent immune recognition. This concept, demonstrated in experimental models,9 can be translated into clinical practice through T-cell recognition of leukemic cells via class I or even by their activation in response to bystander targets, including healthy tissues. It is thus possible to envisage (and should be tested in upcoming clinical studies) that, upon development of nonsevere systemic GVHD and consequent release of proinflammatory stimuli, these patient might recover expression of HLA class II and return to benefit from a proficient GVT response.
Upregulation of inhibitory T-cell ligands
Both genomic HLA loss and downregulation of HLA class II molecules represent strategies used by malignant cells to become “invisible” to donor-derived T cells. However, there is also an alternative modality used by malignant cells to avoid elimination by T cells: conveying an inhibitory signal during the encounter, leaving effectors armed but exhausted. These types of inhibitory interactions, commonly referred as immune checkpoints, have been described to be of fundamental relevance to evade physiological cancer immunosurveillance during the development and progression of many solid tumors, but, with the relevant exception of some subtypes of lymphoma, they appear less involved in the natural biology of hematological malignancies, and especially of leukemias. This picture changes significantly, however, in patients relapsing after chemotherapies,36 and even more dramatically in those relapsing after allo-HCT, where it is possible to document the expression of multiple inhibitory ligands on malignant cells,9,32,37 mirrored by concomitant upregulation of the relative receptors on donor-derived T cells.9,38,39 Although the best characterized of these interactions is the one between PD-1 (on T cells) and PD-L1 (on malignant cells), the landscape of immune checkpoints involved in posttransplant relapse is composite and in continuous expansion (Figure 2, right column). Thus, in a single patient, multiple inhibitory interfaces will possibly overlap, and different patients will present different combinations, warranting an in-depth analysis of each case for identification of potential therapeutic targets. Of interest, T cells that coexpress multiple checkpoints are expectedly also the ones to interact more closely and for the longest time with their target cells; thus, studying the pattern of expression of inhibitory receptors can indirectly help to identify antitumor specificities. Using this approach, it has recently been shown that, in patients with AML with posttransplantation relapse, it is possible to detect coexpression of inhibitory receptors on early differentiated memory T cells residing in the bone marrow (representing the “reservoir” of antigen-specific responses), and that these markers of a deeply dysfunctional and exhausted immune compartment precede by several months the occurrence of relapse.38
From a therapeutic perspective, besides the hurdle of possibly having to block multiple interactions to achieve significant clinical benefit, the use of checkpoint blockade in patients who have received allo-HCT also poses serious concerns about the risk of unleashing alloreactive T cells against healthy tissues. Indeed, the first clinical reports on the use of this approach to treat posttransplantation relapse have shown promising signs of efficacy,40,41 but (especially with anti–PD-1 antibodies) they also have shown a significant rate of severe acute GVHD.42 It should also be noted that many of the inhibitory ligands described to date, including PD-L1, can be potently induced by proinflammatory stimuli, including interferon-γ.43 This means that, in patients relapsing through this mechanism (opposite to those with downregulated HLA class II expression), induction of GVHD will not rescue, and may worsen, the immune phenotype.
Finally, it should be considered that the width of the T-cell repertoire transferred and reconstituted in the patient might correlate significantly with protection against relapse. The recent development of high-throughput TCR sequencing technologies provides the opportunity to analyze in fine detail this important parameter in the interaction between adaptive immune system and tumor, and initial studies have already shown the influence of different GVHD prophylaxes on the reconstituting TCR repertoire44 and a narrow spectrum of TCRs in the exhausted bone marrow T cells of relapsing patients.38
Integrating clonal evolution and immune changes
Even if for the sake of simplicity they were presented separately, changes in the mutational pattern and the immunogenicity of malignant cells not only can occur concomitantly and exploit the same mechanisms but also can actually coincide. One example is provided by FLT3-ITD mutations, which, as mentioned above, are often selectively enriched at posttransplantation relapse.8,9 A recent study highlighted that, in addition to its well-established oncogenic effect, FLT3-ITD can dampen the ability of myeloid cells to release interleukin-15 (IL-15), one of the most essential cytokines for both early differentiated T cells and natural killer cells. Thus, therapeutic blockade of FLT3-ITD after allo-HCT not only can halt leukemia proliferation but also can induce its release of IL-15, facilitating the GVT effect.45 Similarly, Janus kinase 2 mutations, which also can represent an adjunctive proliferation signal gained by leukemic cells during progression, can promote the expression of PD-L1 on malignant cells and their more differentiated progeny, impairing T-cell activation, metabolism, and cell cycle progression.46 Although not studied in the setting of hematological malignancies, also the most common oncogenic mutations of KRAS have been shown to promote immune escape through a number of mechanisms, including the stabilization of PD-L1 mRNA,47 the induction of its surface expression,48 and the recruitment in the tumor site of myeloid suppressor cells.49
Whereas all of these are examples of oncogenic mutations promoting immune evasion, it is also true that oncogenic and immune drivers can diverge during disease progression. This is exemplified by several patients with HLA loss relapses in whom a less malignant but immunoprivileged clone carrying the HLA CN-LOH eventually outgrew over others with a higher number of oncogenic mutations and that were more prevalent at the time of diagnosis (Sala E, Biavasco F, Bucci G, et al, unpublished data).
Clinical case: reassessment and treatment
A complete disease reassessment was performed at the time of posttransplantation relapse. It provided evidence that the patient’s cytogenetics were unchanged but the mutational pattern was different, with evidence remaining of NPM1 mutA but absence of FLT3-ITD. Immunophenotypic analysis documented no changes in lineage-specific and disease-associated markers, conserved expression of HLA class II, and no significant upregulation of PD-L1. Molecular analysis using the HLA-KMR assay evidenced genomic loss of the incompatible HLA haplotype.
On the basis of these results, the patient received reinduction chemotherapy without addition of FLT3 inhibitors or DLIs (which would have been indicated in the confirmed presence of FLT3-ITD and incompatible HLA haplotype, respectively). Upon achieving second remission, the patient underwent second transplantation from a different haploidentical donor. His cousin was selected as the first choice because she carried the HLA haplotype lost by leukemic cells and was incompatible for the retained haplotype.
Conclusions and future perspectives
This review was aimed at summarizing current knowledge on the best established drivers of posttransplantation relapse, but the full picture is probably much more complex, including not only oncogene-driven or immune-related tumor-intrinsic mechanisms but also tumor-extrinsic mechanisms prompted by the pathological niche microenvirmonment.17 Moreover, data are already emerging on the extent and clinical relevance of epigenetic clonal evolution, which may by far surpass its genomic counterpart.50 Epigenetic therapies, in particular those employing the DNA methyltransferase inhibitors azacitidine and decitabine, can at the same time alter these dynamics and the immunogenicity of tumor cells. Demethylating agents have in fact already been shown to have an impact on the expression of HLA molecules51 and immune checkpoints, including PD-L1,52 and this might at least partly explain the promising results obtained with these drugs when treating posttransplantation relapses.53,54
It is thus of fundamental relevance to develop comprehensive models to understand the key alterations that have promoted recurrence in each specific patient and to translate this information into rationales for personalized therapy. Although it is undeniable that the results of available treatments for relapse have been to date discouraging, it is also true that, for each tested approach (DLIs, promotion of nonsevere GVHD, second transplantation), there has been a small but reproducible subset of exceptional responders. The challenge will be to understand whether, by integrating the expanding knowledge of relapse biology in clinical practice, it will be possible to better allocate available therapeutic options, and possibly integrate them with new approaches, including targeted agents and epigenetic therapies.
Acknowledgments
The author thanks Dr. Cristina Toffalori for drafting the manuscript figures and all members of the Unit of Immunogenetics, Leukemia Genomics and Immunobiology, IRCCS San Raffaele Scientific Institute, Milan, Italy, for critical reading and discussion on the text.
The author acknowledges support from the Italian Ministry of Health (RF-2011-02351998, RF-2011-02348034, and TRANSCAN HLALOSS), the Associazione Italiana per la Ricerca sul Cancro (Start-Up Grant 14162 and IG 22197), and the DKMS Mechtild Harf Foundation (DKMS Mechtild Harf Research Grant 2015).
Correspondence
Luca Vago, Unit of Immunogenetics, Leukemia Genomics and Immunobiology, IRCCS San Raffaele Scientific Institute, via Olgettina 60, 20132 Milan, Italy; e-mail: vago.luca@hsr.it.
