
Abstract
von Willebrand disease (VWD) is the most common autosomal inherited bleeding disorder, with an estimated prevalence of 1 in 1000 individuals. VWD is classified into quantitative and qualitative forms. Diagnosis of VWD is complex and requires (1) a personal history of bleeding symptoms, (2) family history of bleeding or VWD, and (3) confirmatory laboratory testing. There are certain bleeding assessment tools to objectively measure bleeding symptoms in patients that have been shown to correlate with the diagnosis as well as the severity of VWD. Laboratory diagnosis requires at least initially a measurement of von Willebrand factor (VWF) antigen levels, VWF platelet binding activity (VWF:RCo, VWF:GPIbM, and VWF:GPIbR), and factor VIII (FVIII) activity. Additional testing to confirm the specific subtype may include VWF collagen binding activity, low-dose ristocetin VWF-platelet binding, FVIII-VWF binding, VWF multimer analysis, and VWF propeptide antigen. Recent advances have been made regarding some of these assays. Molecular testing in VWD is not found to be useful in “low VWF” or most type 1 VWD cases but may be informative in patients with severe type 1 VWD, type 1C VWD, type 2 VWD, or type 3 VWD for accurate diagnosis, genetic counseling, and appropriate treatment. The diagnostic algorithm for VWD is complex, but advances continue to be made in improving VWF functional assays and diagnostic pathways.
Learning Objectives
Understand advances in clinical evaluation of patients with von Willebrand disease
Understand new diagnostic assays for von Willebrand disease
Clinical case
A 4-year-old female presents with recurrent epistaxis and easy bruisability. She has never had any surgical challenges. There is a possible family history of von Willebrand disease (VWD) in the father, but he does not live with the family. Laboratory evaluation showed mild anemia with a hemoglobin of 9.6 g/dL, normal platelet count, and normal white count. von Willebrand factor antigen (VWF:Ag) was 36 IU/dL, von Willebrand factor (VWF):RCo activity was <10 IU/dL, and von Willebrand factor collagen binding activity (VWF:CB) activity was also low at 15 IU/dL. Multimer evaluation showed loss of high-molecular weight multimers. Considering the discrepant VWF:RCo to VWF:Ag ratio, a more accurate assay to evaluate the platelet binding activity of VWF was evaluated. This showed a VWF:Ag of 32 IU/dL and a VWF:GPIbM activity of 12 IU/dL. A question about type 2A VWD vs type 2B VWD was raised. A ristocetin-induced platelet aggregation (RIPA) and a VWF-platelet binding study were found to be normal. Genetic testing was sent for confirmation starting with sequencing of exon 28 of the VWF gene. The p.D1472H variant was identified. This variant has been shown to cause disproportionately low VWF:RCo activity compared with VWF:Ag but has not shown to be associated with bleeding symptoms. To further evaluate for other potentially implicated variants, other exons were sequenced, and they have been shown to have variants associated with type 2A VWD, including exons 11 to 17, 22, 25 to 28, and 52. A variant in exon 26 was identified, c.3389G>T (Cys1130Phe), which has been previously reported as a pathogenic variant for autosomal dominant type 2A VWD. Thus, this patient was confirmed to have type 2A VWD.
Introduction
VWD is the most common autosomal inherited bleeding disorder, with an estimated prevalence of 1 in 1000 individuals.1 Classification of VWD is currently based on the criteria developed by the VWF subcommittee of the International Society on Thrombosis and Haemostasis (ISTH) and the National Institutes of Health National Heart, Lung, and Blood Institute (NHLBI) VWD expert panel, and these guidelines were published in 2006 and 2007, respectively.1,2 VWD is classified into 3 major categories: partial quantitative VWF deficiency (type 1), complete deficiency (type 3), and qualitative deficiency (type 2). Type 2 is further classified into subtypes defined by defects in multimerization (type 2A), increased platelet binding (type 2B), defects in VWF-platelet binding or VWF:CB (type 2M), or defects in factor VIII (FVIII) binding (type 2N). Type 1 is the most common, accounting for about 85% of VWD, whereas type 3 is the least common, affecting an estimated 1 in 1 million individuals. Type 2 VWD is less common than type 1. Diagnosis of VWD includes (1) assessment of history of bleeding symptoms, (2) assessment of family history of bleeding or VWD, and (3) confirmatory laboratory testing.3
Bleeding assessment tools
VWD is characterized by increased or excessive mucocutaneous bleeding manifesting as epistaxis; easy bruising; prolonged bleeding from minor wounds; oral cavity bleeding; bleeding after surgery, dental extraction, or childbirth; and heavy menstrual bleeding. The most severely affected individuals experience musculoskeletal bleeding, including hemarthrosis and muscle hematomas. To objectively quantify the severity of bleeding symptoms, several bleeding scores have been devised. The ISTH has developed a validated Bleeding Assessment Tool (BAT) for evaluation of patients with VWD. Normal ranges of the ISTH BAT have been established for adult patients (males and females) and pediatric patients.4 The ISTH BAT scores have been shown to correlate with the severity of VWD. ISTH BAT scores are higher in type 3 VWD than in type 2 or type 1, and they are higher in children with severe VWD (VWF:Ag < 10 U/dL) than children with moderate VWD (VWF:Ag = 10-30 U/dL).5 Bowman et al6 published the Pediatric Bleeding Questionnaire (PBQ), which has pediatric-specific bleeding symptoms, such as umbilical stump bleeding, cephalohematoma, postcircumcision bleeding, postvenipuncture bleeding, and macroscopic hematuria. The high negative predictive value of the PBQ suggests that this tool could be a valuable addition to the evaluation of children with suspected inherited bleeding disorders. Similar results have been obtained in prospectively studied adults screened for suspected bleeding tendency.7 One limitation of these BATs is that they require expert administration, which can be problematic from a resource perspective, especially in a busy clinical setting, and it is a barrier to more widespread use of such tools. A self-administered BAT was recently validated as a screening tool for VWD and shown to correlate well with expert-administered ISTH BAT scores, making it a lucrative option to use in a busy clinic setting.8 Additionally, these tools are limited by the fact that pediatric patients may not have had as many challenges to experience significant bleeding symptoms. A pediatric self-BAT has recently been validated.9 Menorrhagia-specific questionnaires are also available for evaluation of heavy menstrual bleeding in many patients with VWD. These are the Pictorial Bleeding Assessment Chart (PBAC) and Phillip scores. The Phillip tool combined with the PBAC score >100 has a sensitivity of 95% for inherited bleeding disorders, especially VWD.10
Diagnostic criteria for VWD
Clinical laboratory testing for VWD initially includes measurement of at least VWF:Ag, VWF-platelet binding activity (VWF:RCo, VWF:GPIbM, and VWF:GPIbR), and FVIII level (Table 1).3 Additional testing may be indicated based on the initial test results, including low-dose ristocetin VWF-platelet binding, VWF multimers, von Willebrand factor propeptide (VWFpp) level, and VWF:CB. Ratios of VWF functional activity (VWF:RCo and VWF:CB), FVIII level, or VWFpp level to VWF:Ag are used in identification of VWD subtype. However, caution must be used when assessing these ratios at very low VWF:Ag levels, because variability of values at the lower end of the analytical range may result in an unreliable ratio. In addition, evaluation of other family members for bleeding symptoms as well as laboratory testing for VWD in those with bleeding symptoms may be beneficial.
Typical laboratory test results for VWD subtypes
| Normal | Type 1 | Type 1C | Type 2A | Type 2B* | Type 2M | Type 2N | Type 3 | |
|---|---|---|---|---|---|---|---|---|
| VWF:Ag | N | ↓↓ | ↓↓ | N, ↓, or ↓↓ | N, ↓, or ↓↓ | ↓ or ↓↓ | N or ↓ | Absent |
| VWF:RCo/VWF:Ag, VWF:GPIbM/VWF:Ag, VWFGPIbR/VWF:Ag | N | N | N | ↓↓ | ↓ or ↓↓ | ↓↓ | N | N/A |
| FVIII/VWF:Ag | N | N | N | N | N | N | ↓↓ | N/A |
| LD-RIPA | None | None | None | None | ↑↑ | None | None | None |
| Platelet count | N | N | N | N | N or ↓ | N | N | N |
| VWF multimers | N | N | N | ↓HMW | ↓HMW | N | N | Absent |
| VWFpp/VWF:Ag | N | N | ↑ or ↑↑ | N or↑ | N or ↑ | N | N | N/A |
| VWF:CB/VWF:Ag | N | N | N | ↓↓ | ↓ or ↓↓ | N or ↓ | N | N/A |
| Normal | Type 1 | Type 1C | Type 2A | Type 2B* | Type 2M | Type 2N | Type 3 | |
|---|---|---|---|---|---|---|---|---|
| VWF:Ag | N | ↓↓ | ↓↓ | N, ↓, or ↓↓ | N, ↓, or ↓↓ | ↓ or ↓↓ | N or ↓ | Absent |
| VWF:RCo/VWF:Ag, VWF:GPIbM/VWF:Ag, VWFGPIbR/VWF:Ag | N | N | N | ↓↓ | ↓ or ↓↓ | ↓↓ | N | N/A |
| FVIII/VWF:Ag | N | N | N | N | N | N | ↓↓ | N/A |
| LD-RIPA | None | None | None | None | ↑↑ | None | None | None |
| Platelet count | N | N | N | N | N or ↓ | N | N | N |
| VWF multimers | N | N | N | ↓HMW | ↓HMW | N | N | Absent |
| VWFpp/VWF:Ag | N | N | ↑ or ↑↑ | N or↑ | N or ↑ | N | N | N/A |
| VWF:CB/VWF:Ag | N | N | N | ↓↓ | ↓ or ↓↓ | N or ↓ | N | N/A |
HMW, high molecular weight; LD-RIPA, low-dose ristocetin-induced platelet aggregation; N, normal; ↓, reduced; ↓↓, markedly reduced; ↑, increased; ↑↑, markedly increased.
Platelet-type VWD has a similar laboratory phenotype.
Type 3 VWD is characterized by undetectable VWF protein and markedly reduced FVIII level (Table 1). In type 1 VWD, VWF:Ag level is low, and VWF-platelet binding activity is reduced in parallel. FVIII level may be reduced secondary to reduced VWF level, and VWF multimer structure should be essentially normal, with no significant decrease in the large VWF multimers. The diagnostic VWF cutoff levels for type 1 diagnosis have been debated for many years.11 Previous NHLBI guidelines recommended a cutoff of <30 IU/dL; however, this cutoff varies widely in practice.2 Some use the lower limit of normal (∼50 IU/dL), and other reports use <40 IU/dL. Patients with intermediate plasma VWF levels (30-50 IU/dL) have also been categorized as “low VWF levels.” A recent study by Lavin et al12,13 concluded that low VWF levels may be associated with significant bleeding in patients owing to reduced VWF synthesis/secretion. Diagnostic cutoffs for type 1 VWD and “low VWF” remain to be delineated.
Type 2 VWD encompasses abnormalities in VWF function. Initial laboratory testing in type 2A and type 2B VWD may be similar with normal or reduced VWF:Ag level, markedly reduced VWF-platelet binding activity (eg, decreased VWF:RCo/VWF:Ag), normal to reduced FVIII level, and loss of high-molecular weight multimers (Table 1).2 Patients with type 2B often have thrombocytopenia that is not observed in type 2A because of variants that cause increased VWF-platelet binding and clearance of large multimers.14 Abnormally increased RIPA at low ristocetin concentration is diagnostic for type 2B VWD. Genetic testing of the VWF gene may also be useful in discriminating type 2A from 2B as discussed in more detail below. The majority of type 2M VWD includes variants with decreased VWF binding to platelets not caused by abnormal multimer structure (eg, decreased VWF:RCo/VWF:Ag). Initial test results may be similar to type 2A, with discordant VWF:Ag and VWF-platelet binding activity; however, type 2M subjects have normal VWF multimer structure. A less common and less well-known variation of type 2M includes subjects with VWF collagen binding defects (Table 1). These subjects have reduced VWF:CB/VWF:Ag ratio and normal VWF multimers. Type 2N VWD is caused by variants that impair FVIII binding, causing low FVIII levels. The diagnostic hallmark for type 2N VWD is markedly discordant VWF:Ag and FVIII level as identified by a markedly reduced FVIII/VWF:Ag ratio. Discrimination of type 2N from hemophilia A may require the assay of FVIII-VWF binding, which is becoming more readily available in clinical or reference laboratories. Alternatively, genetic testing may be used to confirm type 2N VWD.
Assays of VWF-platelet binding activity
The classic assay for assessing the ability of VWF to bind to platelets is VWF:RCo activity where the antibiotic ristocetin is used to induce a conformational change, exposing the VWF A1 domain to allow platelet GPIbα binding (Figure 1). VWF:RCo has been used for decades, and most data correlating VWF levels and treatment outcomes have used VWF:RCo. However, the VWF:RCo assay has a high coefficient of variation and poor sensitivity, making it difficult to assess patients with very low VWF levels (<20 IU/dL).15 VWF:RCo measures not only the ability of VWF to bind GPIbα but also, VWF-ristocetin binding. False positive results may occur in subjects with the p.D1472H VWF variant that is common in African Americans and less common in other subjects.16 Other variants in the VWF-ristocetin binding region may impact the VWF:RCo assay in a similar manner.
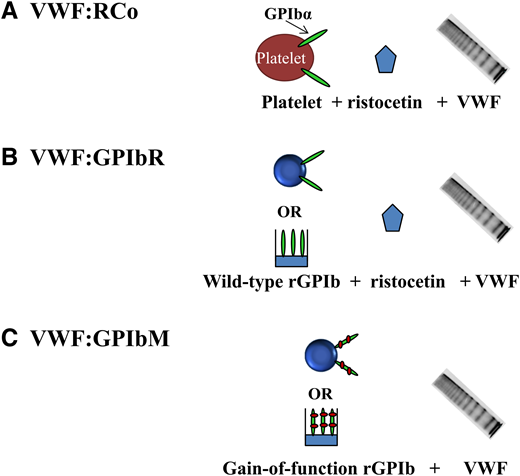
Platelet-dependent VWF activity assays. (A) VWF:RCo includes “traditional” assays that use ristocetin to induce VWF binding to platelets. (B) VWF:GPIbR includes assays based on ristocetin-induced binding of VWF to recombinant wild-type GPIbα fragments tethered to either latex particles or an enzyme-linked immunosorbent assay (ELISA) plate. (C) VWF:GPIbM assays are based on spontaneous binding of VWF to recombinant gain-of-function GPIbα fragments tethered to either latex particles or an ELISA plate
Platelet-dependent VWF activity assays. (A) VWF:RCo includes “traditional” assays that use ristocetin to induce VWF binding to platelets. (B) VWF:GPIbR includes assays based on ristocetin-induced binding of VWF to recombinant wild-type GPIbα fragments tethered to either latex particles or an enzyme-linked immunosorbent assay (ELISA) plate. (C) VWF:GPIbM assays are based on spontaneous binding of VWF to recombinant gain-of-function GPIbα fragments tethered to either latex particles or an ELISA plate
A recently developed assay (VWF:GPIbR) uses recombinant GPIbα fragments tethered to microparticles, eliminating the use of whole platelets (Figure 1).15,17 VWF:GPIbR is less variable and more sensitive than VWF:RCo, but it has excellent correlation with VWF:RCo, and also, it is commercially available in Europe and Canada.15 Boender et al18 recently reported that VWF:GPIbR is unaffected by the common p.D1472H VWF variant, unlike VWF:RCo. Another recently developed assay, VWF:GPIbM, uses recombinant GPIbα fragments with 2 gain-of-function variants to induce spontaneous binding of VWF without ristocetin (Figure 1).19,20 Although VWF:GPIbM eliminates the nonphysiologic ristocetin, it does introduce a nonphysiologic mutant GPIbα receptor. VWF:GPIbM is reported to have an excellent coefficient of variation, a more sensitive lower limit of detection, and excellent correlation with VWF:RCo.15,21 VWF:GPIbM is available commercially on an automated platform in Canada and Europe, whereas an enzyme-linked immunosorbent assay version is available in the United States through Versiti, Blood Center of Wisconsin. Because ristocetin is eliminated, VWF:GPIbM results are unaffected by the p.D1472H VWF variant.18,20
In a recent study by Boender et al,18 VWF:RCo, VWF:GPIbR, and VWF:GPIbM assay results were compared using a well-defined cohort of VWD subjects. They found excellent correlation between assays but discrepant classifications for many VWD patients. VWF:RCo misclassified up to 18% of patients and half of type 2B subjects. VWF:GPIbR was more sensitive and accurate in classifying the majority of patients, whereas VWF:GPIbM was the most precise but misclassified several of genotypic type 2A, type 2B, and type 3 patients. VWF:GPIbM may be increased in type 2B subjects, potentially because of the combination of increased platelet affinity of type 2B VWF with the gain-of-function mutant GPIbα protein used. Although this study did have some limitations, including differences in calibration between assays as well as the use of a single commercially available assay of each type, it did reveal that VWF:RCo has been surpassed by VWF:GPIbR and VWF:GPIbM assays in terms of precision, sensitivity, and diagnostic accuracy.
Assay of VWFpp
The assay of VWFpp level can also be useful in the diagnosis of VWD, particularly in defining VWD subtypes.22-24 The VWFpp level is used as a surrogate marker of VWF synthesis and secretion. VWFpp is most useful in identifying the subset of type 1 VWD subjects with very low VWF:Ag owing to increased clearance of VWF from plasma, which is commonly referred to as type 1C.22,25,26 These patients have a markedly reduced VWF half-life, which is as short as 1 to 3 hours after desmopressin administration, in contrast to the normal 8- to 12-hour half-life. By definition, healthy individuals have 1 U each of VWF and VWFpp in plasma, and they should have a steady-state ratio of <3.0. In type 1C patients, VWFpp is assumed to have a normal half-life, with substantially reduced VWF half-life resulting in an increased VWFpp/VWF:Ag ratio (>3.0). Identification of type 1C patients has implications for treatment strategies. These patients may release VWF after desmopressin treatment, but the released VWF will be quickly cleared from plasma. Increased clearance of VWF also occurs in some types of acquired VWD and in type 2B and some type 2A patients as identified by an elevated VWFpp/VWF:Ag ratio.27
Sanders et al23 studied a large group of type 1, type 2, and type 3 VWD subjects using VWFpp/VWF:Ag and FVIII/VWF:Ag ratios to define the pathological mechanism (reduced VWF survival, reduced VWF secretion, or both) causing the VWD phenotype. This study highlighted the utility of VWFpp levels in identifying true type 3 VWD patients, because some patients had severely reduced VWF:Ag with detectable or normal VWFpp level, indicating reduced VWF survival rather than an absence of VWF synthesis. The reclassification of these patients has implications for treatment options. Where type 3 VWD patients do not produce VWF, type 1C subjects should have relatively normal endothelial cell Weibel–Palade body and platelet α-granule VWF stores. The VWF released after desmopressin administration, although quickly cleared from plasma, may be sufficient to achieve hemostasis in minor bleeding situations.28 Type 3 patients will not respond to desmopressin and require VWF replacement therapy. The VWFpp assay is not routinely included in the diagnostic workup for VWD, and it is available in few diagnostic laboratories; however, it may be a valuable addition to the diagnostic panel, particularly in patients with markedly reduced VWF:Ag levels. Although this assay provides little additional information in type 2 VWD, it has utility in discriminating between type 3, type 1, type 1C, and some types of acquired VWD, with implications for therapeutic treatment.
VWF:CB assay
Yet another VWF function is to bind to exposed collagen at the site of vascular injury, and it requires an assay to specifically examine VWF-collagen interaction. VWF has been shown to bind to several different types of collagen.29 The VWF A3 domain binds to type 1 and type 3 collagen, whereas type 4 and type 6 collagen bind through the VWF A1 domain. Most VWD diagnostic panels typically do not include any assessment of VWF:CB, and the clinical laboratories that do perform this testing use type 1 or type 3 collagen. Currently, the assay of type 4 or type 6 collagen binding is not clinically available.
VWF collagen binding is dependent on the presence of high-molecular weight VWF multimers, and patients with defective VWF multimers (type 2A or type 2B) have reduced VWF:CB (Table 1).30 Flood et al31 showed that collagen binding provides a sensitive screen for variant VWD, and VWF:CB serves as a surrogate for the presence of high-molecular weight VWF multimers. Patients with normal multimer structure should have a VWF:CB/VWF:Ag ratio of ∼1.0. Patients with type 2A or type 2B VWD with abnormal VWF multimers have reduced VWF:CB/VWF:Ag ratio. The sensitivity of VWF:CB for multimer abnormalities was 97% to 100% for healthy controls and type 1, type 2A, and type 2B VWD depending on the VWF:CB/VWF:Ag cutoff used. Other investigators have reported on the utility of VWF:CB/VWF:Ag to distinguish VWF multimer distribution.30,32,33 VWF:CB may prove to be more efficient and potentially less costly than VWF multimer analysis. However, VWF multimer analysis by classical electrophoresis is likely warranted to confirm VWD subtype after initial VWD diagnosis.
Assessment of VWF:CB activity may have another role in identifying patients with collagen binding defects falling under the category of type 2M VWD. Assays using type 1 or 3 collagen will detect the rarer specific VWF A3 domain collagen binding variants.34 VWF A1 domain collagen binding variants are more common and can be identified using type 4 or 6 collagen, but this assay is not currently clinically available.35 Both A1 and A3 domain collagen binding variants have been reported to be associated with an increased bleeding score compared with similar subjects without an identified collagen binding variant. Addition of VWF:CB activity to the VWD diagnostic pathway may serve a dual role in evaluating VWF multimer structure and identifying a possible collagen binding defect.36
Genetic testing in VWD
Genetic testing for VWD is not common practice and should be used in specific cases, where it may have implications for diagnosis, management, or counseling. Genetic testing in type 1 VWD is not very informative, because an average of 62% of patients with type 1 VWD have an identifiable sequence variant (SV); however, for patients with VWF < 30 IU/dL, the detection rate of identifiable SVs is ∼80%.37 Genetic testing in type 2 VWD may help to confirm the molecular diagnosis and the specific subtype to aid in patient management. It can be particularly helpful in diagnosing patients with type 2N VWD vs mild hemophilia A, differentiating platelet-type VWD from type 2B VWD, and confirming diagnosis of type 2M and type 2A VWD. In patients with type 3 VWD, genetic testing may be helpful for accurate diagnosis (misdiagnosis of some severe type 1 or 2 patients), to determine risk of alloantibody development (more commonly seen with large deletions), and for genetic counseling and risk determination for other family members (parents or offspring of patients with type 3 VWD may have type 1 VWD or be phenotypically normal depending on the variant type).38
There are certain limitations of genetic testing that may complicate genetic VWD diagnosis. VWF is a large gene consisting of 52 exons, with a highly homologous partial pseudogene corresponding to exons 23 to 34. This pseudogene makes sequencing and interpretation particularly difficult. Utilizing a diagnostic laboratory with significant VWF gene sequencing experience is essential. Additionally, VWF gene is highly polymorphic, with significant variability in normal individuals. Many variants previously classified as pathogenic have been found in healthy people, questioning their significance.39 Therefore, DNA changes must be correlated with functional VWF studies for accurate diagnosis.16 For example, a common single-nucleotide polymorphism (SNP) in exon 28 of VWF, p.D1472H, has been associated with a significantly low VWF:RCo/VWF:Ag ratio; however, it does not have any functional significance, and it does not cause disease.16 Identifying this SNP may be informative in those patients who are misdiagnosed as type 2M VWD, particularly those of African-American descent. The sequencing of the entire VWF coding region also has limitations owing to its high costs, but next generation sequencing (NGS) has emerged as an alternative to overcome this limitation.40,41 NGS technologies can be economically advantageous when the sequencer runs at full capacity, allowing a large number of samples to be analyzed to reduce cost. Also, a tiered approach is often found useful for genetic diagnosis of VWD, because many variants are clustered in specific regions, making it less cumbersome and cost effective to analyze these regions.
DNA sequencing by Sanger sequencing or NGS cannot detect copy number variants (CNVs), including deletions and duplications, which cause ∼10% of genetic diseases. Christopherson et al42 showed that large deletions were identified in 12% of their VWD cohort (including VWD type 1, type 1C, and type 3). Array comprehensive genomic hybridization is a sensitive and powerful method to identify CNV in the VWF gene and should be considered when attempting to determine the underlying genetic cause of type 1 VWD patients with VWF:Ag levels <30 or type 3 VWD patients when an SV is not identified by VWF sequencing.
Case follow-up
Our patient with significantly abnormal VWF studies was confirmed to have type 2A VWD. The patient had a suboptimal response to inhaled desmopressin. She now receives desmopressin for minor bleeding and VWF concentrate for more significant bleeding or surgical prophylaxis.
Correspondence
Sandra L. Haberichter, Diagnostic Laboratories and Blood Research Institute, Versiti, 638 N 18th St, PO Box 2178, Milwaukee, WI 53201; e-mail: shaberichter@versiti.org.
