
Abstract
Chronic lymphocytic leukemia has a highly variable disease course across patients, thought to be driven by the vast inter- and intrapatient molecular heterogeneity described in several large-scale DNA-sequencing studies conducted over the past decade. Although the last 5 years have seen a dramatic shift in the therapeutic landscape for chronic lymphocytic leukemia, including the regulatory approval of several potent targeted agents (ie, idelalisib, ibrutinib, venetoclax), the vast majority of patients still inevitably experience disease recurrence or persistence. Recent genome-wide sequencing approaches have helped to identify subclonal populations within tumors that demonstrate a broad spectrum of somatic mutations, diverse levels of response to therapy, patterns of repopulation, and growth kinetics. Understanding the impact of genetic, epigenetic, and transcriptomic features on clonal growth dynamics and drug response will be an important step toward the selection and timing of therapy.
Learning Objectives
Identify genetic and phenotypic characteristics that impact clonal growth dynamics
Understand the latest research methodologies used to describe clonal evolution
Introduction
Tumor development, progression, and resistance to therapy are steps of a dynamic evolutionary process that applies across all cancer types. A byproduct of evolution is the presence of numerous subpopulations within a tumor that can be unique in their molecular characteristics, growth kinetics, and response to therapy.1,2 This process has notably been observed in chronic lymphocytic leukemia (CLL), a B-cell malignancy thought to originate from monoclonal B-cell lymphocytosis that accumulates in the blood and lymphoid organs.3 Over the past decade, large-scale studies leveraging next-generation sequencing (NGS) have uncovered vast inter- and intratumoral heterogeneity in CLL.2,4-6 Clinically, CLL also has a diverse presentation, with patients progressing at variable rates with a range of responses to therapy. Despite the introduction of novel potent targeted therapies to the clinic in the past 5 years, including Bruton tyrosine kinase (BTK) inhibitors (such as ibrutinib and acalabrutinib), phosphatidylinositol 3-kinase inhibitors (including idelalisib, duvelisib, and copanlisib), and B-cell lymphoma 2 (BCL2) inhibitors (venetoclax), the majority of patients remain with disease.7-9
Recent findings have linked molecular heterogeneity in CLL to diverse clonal dynamics and therapeutic responses.1,2,7,8 Using the following clinical cases, we highlight the potential impact of these insights on the prognostication of disease and the selection and timing of therapy.1 On the basis of each patient’s initial cytogenetics, what can we expect of their respective prognoses and responses to treatment? What additional information would we need to better predict their clinical courses?
Clinical case 1
A 51-year-old man has newly diagnosed CLL and Rai stage I disease. His blood examination at diagnosis shows a white blood cell (WBC) count of 23.3, and he is identified as having mutated immunoglobulin heavy chain (IgH) disease with del(13q).
Clinical case 2
A 51-year-old man has newly diagnosed CLL and Rai stage I disease. His blood examination at diagnosis shows a WBC count of 14.8, and he is identified as having unmutated IgH disease with del(13q) and a positive ZAP70 status.
Tumor heterogeneity in CLL
Clonal evolution, one of the major factors underlying the intractability of cancer, is driven by underlying intratumoral heterogeneity. Genetic and epigenetic factors can together influence a cell’s phenotype, growth rate, and response to environmental pressures. These resulting dynamics in turn help shape the clonal makeup of tumors and explain the heterogeneity often seen in tumor progression in the absence and presence of therapy. Clonal patterns described across cancers can include clonal equilibrium, in which the relative abundance of each subclone is maintained in a mixed population (Figure 1A), as well as clonal competition, whereby heritable (often genetic) alterations result in differential fitness that influences the prevalence of each clone in a population (Figure 1B). With regard to clonal competition, distinct evolutionary patterns have been observed. Linear evolutionary trajectories, for example, involve a progeny clone replacing its parent clone in a full selective sweep and tend to occur during particularly stringent conditions (ie, therapeutic treatment). Branched trajectories, in which multiple subclones coexist in the same tumor and compete for dominance, can occur during passive or active evolutionary settings but unfold at different tempos (gradual vs punctuated) accordingly.2,3,10-13 The extent to which these evolutionary patterns are observed across cancers has been well-documented in recent years, particularly with the advent of NGS.
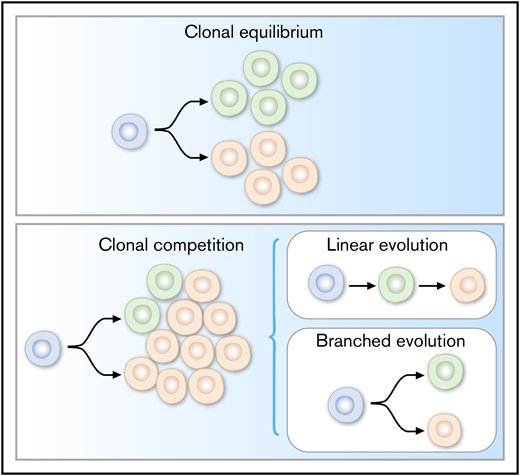
Evolutionary mechanisms (linear and branched) underlying clonal dynamics in cancer.
Evolutionary mechanisms (linear and branched) underlying clonal dynamics in cancer.
CLL was among the first cancers characterized by modern sequencing methods for which clonal evolution has been delineated. This is due in large part to its relative indolence (allowing longitudinal sampling over the course of disease), easy accessibility to high-purity samples via serial venipuncture, and a highly variable clinical course that allows mapping of molecular characteristics to prognosis and clinical outcomes. The earliest reports of genetic heterogeneity in CLL from karyotyping, fluorescence in situ hybridization, and single-nucleotide polymorphism arrays collectively uncovered recurrent copy number alterations, most commonly involving deletions of chromosomes 11q and 17p and trisomy of chromosome 12 (all of which associate with more aggressive disease) and focal deletion of chromosome 13q in more than half of all patients (which is associated with lower risk).12,14,15 Relevant genes within the affected minimally deleted regions of these cytogenetic abnormalities include ATM, BIRC3, TP53, and miR-15a/16. Sequential fluorescence in situ hybridization and chromosome banding analyses over the course of disease progression also provided some of the first insights into clonal evolution at the chromosomal level, revealing that acquisition of chromosomal aberrations over time correlate with IgH mutation status and poor outcomes.16,17
Since then, the advent and decreasing costs of NGS have made large-scale sequencing efforts a tractable approach for studying tumor heterogeneity. Whole-genome and whole-exome sequencing (WGS/WES), for example, have been applied to identify novel coding and noncoding somatic mutations in thousands of patients with CLL worldwide.4,5,18,19 Far from identifying a universal genetic driver, these studies have established that only a small number of genes in CLL are mutated in 10% to 15% of cases (as reviewed by Lazarian et al), whereas a larger number of genes are mutated in <5% of patients.20 In aggregate, these driver genes have implicated a number of key pathways in CLL biology, including DNA damage, RNA processing, signaling pathways, and chromatin remodeling, several of which have been functionally characterized either alone or in relevant combinations through CLL cell lines and mouse models.21-25 The constellation of mutations present within a tumor can be considered a historical record of its evolutionary trajectory and can be used to deduce a temporal order of genomic events. Driver mutations that are clonal likely occurred early in disease and form the trunk of its somatic evolutionary tree (ie, del[13q], tri[12], MYD88), whereas subclonal mutations most commonly present in a small population of leukemic cells and represent later events (ie, TP53, ATM, MGA, BIRC3).4,26 The abundance of each subclonal mutation can be used to infer subclonal hierarchy within the tumor’s phylogenetic tree, allowing estimates of clonal shifts over time. As an analytic strategy, the approach of determining whether a mutation is considered “clonal” or “subclonal” based on the analysis of a single sequenced sample depends on the depth and purity of the sequencing data. Recent studies have demonstrated that the analysis of serial samples from the same patient can greatly increase the sensitivity and confidence in the detection of subclonal mutations.1,27
CLL heterogeneity is not restricted to somatic mutations. Epigenetic mechanisms, such as DNA methylation, chromatin remodeling, and posttranslational histone modification, also contribute to tumor diversity and play a role in cancer evolution.28 Epigenetics, which allow cells to switch between different biological states, can result in differential expression profiles that affect genome stability. This plasticity allows greater adaptability to environmental stressors, including therapy and immune editing. The epigenetic landscape of CLL, studied through analysis of single gene promoters, genome-wide arrays (Illumina 450k), and sequencing (bisulfite sequencing), is relatively stable over time and across resting and proliferative compartments and shares common features with normal B-cell differentiation.29-31 Despite this stability, greater methylation heterogeneity and locally disordered methylation have been described in CLL cells as compared with normal CD19+ B cells. Methylation heterogeneity correlates with genetic subclonal complexity, transcriptional variation, and poor clinical outcome, suggesting that methylation patterns coevolve alongside genetic aberrations as a result (or cause) of genetic instability and provide enhanced potential for alternative evolutionary trajectories.32-36 Furthermore, mutations affecting histone-modifying enzymes and chromatin remodelers have been described in CLL, including EZH2 (catalyzes H3K27 trimethylation and results in transcriptional silencing), SETD2 (responsible for the H3K36me3 histone mark that associates with actively transcribed regions), and ARID1A and CHD2 (chromatin remodelers identified in 2% and 5% of patients with CLL, respectively).28,35,37 Epigenetic heterogeneity has been associated with aggressive features and shorter time to first treatment.36,38
The extent to which epigenetic modifications occur independently of evolutionary changes in DNA is an open question in CLL, and across cancers, in general. Possible interactions between genetic and epigenetic forces in tumor evolution include (1) simultaneous acquisition, whereby a novel mutation in a cancer driver gene is acquired that fundamentally alters the biology of a cell, involving changes to the epigenome; and (2) stepwise acquisition, whereby there first exists a low level of epigenetic instability producing variation within the tumor population that is propagated to all subclones as novel driver mutations arise.33 In CLL, intratumoral methylation heterogeneity in the presence of subclonal mutations is linked to greater overall heterogeneity later in disease, thereby implicating a link between genetic and epigenetic evolution.36
Approaches to studying clonal dynamics in CLL
Alongside the molecular characterization of CLL, numerous methodologies have been employed to link dynamic changes in the genetic and epigenetic features of individual cancers to their phenotype. These approaches largely rely on longitudinal sampling of individual CLLs, which provides sequential snapshots of subclonal composition and which, coupled with measurements of overall tumor burden, can be used to infer clonal fitness and growth kinetics.1,2,4,13 For CLL, estimates of tumor burden can be readily gained from sampling of the absolute lymphocyte count, available from routine peripheral blood (WBCs) measurements. Likewise, for patients with substantial lymph node involvement, tumor burden can be estimated through radiologic imaging and tumor measurements. Altogether, these calculations can contribute to the understanding of the clonal composition of CLL across different organs and the genetic contributors of this localization (Figure 2A). Summarized below are 4 existing and developing approaches that have been applied to the study of CLL clonal kinetics.

Approaches to studying clonal dynamics in CLL. (A) Serial blood draws coupled with volumetric measurements can help monitor CLL progression and contribute to estimations of growth kinetics. (B) Serial samples can increase the sensitivity of clonal detection as compared with depth of sequencing (reprinted from Gruber et al1 with permission). (C) WES, mathematical modeling, and single-cell RNA sequencing can individually and in combination provide insight into determinants of clonal dynamics. Novel lineage-tracing tools are increasingly being leveraged to study clonal dynamics and evolution across cancers (left panel reprinted from Gruber et al1 with permission; top right panel reprinted from Guieze et al66 with permission; bottom right panel reprinted from Trapnell et al44 with permission).
Approaches to studying clonal dynamics in CLL. (A) Serial blood draws coupled with volumetric measurements can help monitor CLL progression and contribute to estimations of growth kinetics. (B) Serial samples can increase the sensitivity of clonal detection as compared with depth of sequencing (reprinted from Gruber et al1 with permission). (C) WES, mathematical modeling, and single-cell RNA sequencing can individually and in combination provide insight into determinants of clonal dynamics. Novel lineage-tracing tools are increasingly being leveraged to study clonal dynamics and evolution across cancers (left panel reprinted from Gruber et al1 with permission; top right panel reprinted from Guieze et al66 with permission; bottom right panel reprinted from Trapnell et al44 with permission).
Bulk genomic analysis
Although CLL, like other blood malignancies, has a much lower mutational burden than solid tumors, we previously established that with even an average of 20 coding mutations per sample, it is feasible to reconstruct clonal architecture and likely phylogenies from an individual sample.39 We recently demonstrated that with increasing number of serial samples, increased sensitivity is gained for detection of these subclonal events (Figure 2B), and that for CLL, the inferences drawn from bulk WES of CLL samples were highly similar to those drawn from WGS performed on the same samples, indicating the robustness of current inference tools for analyses of WES data. With samples collected over 2 time points, it becomes feasible to determine whether clones are contracting vs expanding; with serial sampling of 3 or more time points, greater confidence is gained not only in the building of phylogenetic trees but also for determining clonal dynamics and patterns of growth over time. Recently developed computational tools such as PhylogicNDT can statistically model such trajectories to infer the order of clonal driver events, subclonal populations of cells and their phylogenetic relationships, and overall clonal dynamics in a mixed population.27 Given the limitations of bulk analysis when making clonal inferences or studying transcriptional profiles, single-cell RNA-sequencing analysis will be of increasing interest to the field of clonal dynamics (see section below).
Mathematical modeling
For CLL, mathematical modeling can predict the contribution of genetics to clonal dynamics by using WBC counts or volumetric measurements as a proxy of tumor burden (Figure 2A). Although measurement across 2 time points can provide insight into the clonal diversity and general fitness of subclones, serial measurements over time can highlight the growth patterns and kinetics of bulk tumors and their constituent subclones. By calculating theoretical doubling, it is feasible to define the likelihood of whether individual leukemias display exponential growth kinetics or, rather, a pattern of logistic growth, in which leukemia samples attain carrying capacity, or the maximum number of cells that can be sustainably supported, given the space and resources available (Figure 2C). Integration of genomic information with growth pattern modeling can enable not only the analysis of global tumor growth kinetics but also the tracking of the growth of individual subclones and therefore, potentially, the dynamic interaction between distinct subclones within the same tumor population. Komarova et al led some of the earliest efforts to generate theoretic models using kinetic parameters of CLL (including volumetric changes in lymph node and spleen sizes derived from serial computed tomographic scans and changes in serial blood lymphocyte counts) to predict the evolutionary dynamics of ibrutinib-resistance mutations and the duration of therapeutic efficacy.40 These studies proposed that despite patient-to-patient variation in the division and death rate of CLL cells comprising their malignant cell mass, knowledge of the growth kinetics of resistant clones can inform whether clones involved in relapse are present before therapy and the likely duration of response to therapy. Subsequent studies using deuterated “heavy” water to metabolically label the DNA of proliferating CLL cells in vivo for the study of CLL kinetics (proliferation and death rates) in patients conformed with theoretical data from mathematical modeling, further affirming mathematical modeling as a viable approach for the study of clonal dynamics.41 Recently, application of this type of analysis to real-world data of patients with CLL has been described (see section below).
Single-cell analysis of clonal characteristics
Single-cell technologies have recently emerged as a powerful tool to probe the characteristics of individual cells within a heterogeneous population. Serial molecular profiling of CLLs at the single-cell level over time can provide a granular view of the determinants of clonal fitness. Somatic mutation analysis of DNA from single cells from a sample by single-cell WGS, for example, can provide a comprehensive snapshot of the subclonal composition of a population by definitively identifying the assortment of mutations present in individual clones, but until recently, this was limited by low throughput and high cost. Targeted sequencing panels and droplet digital polymerase chain reaction have served as alternative methods for the DNA analysis of single cells (example in Figures 4D and 4E). Single-cell reduced-representation bisulfite sequencing allows single-cell analysis of genome-wide methylation profiles on a single-nucleotide level.36,42 Single-cell assay for transposase-accessible chromatin using sequencing, which enables the study of chromatin accessibility and cancer-specific transcriptional regulatory networks in individual cells, has yet to be applied to CLL but would likely provide key insights regarding epigenetic reprogramming. Finally, single-cell RNA sequencing is an increasingly popular technique for studying changes in gene expression over time in CLL and is likely to bring many novel and clone-resolved insights into drug response and resistance mechanisms in the near future.43 New computational analysis pipelines have also been developed, such as Monocle, an unsupervised algorithm that can harness single-cell variation to order cells by progress through temporal processes such as differentiation. Monocle can theoretically be applied to serially acquired single-cell data to resolve the transcriptional and clonal dynamics of relapse (Figure 2C).44 Other approaches to analyzing cellular dynamics across temporal processes include the estimation of cell-specific RNA velocity, or the time derivative of the gene expression state, by distinguishing between spliced and unspliced mRNAs in single-cell RNA sequencing data. Application of this tool to the study of therapeutic response in cancer could reveal the rate and direction of change of cellular transcriptomes across thousands of individual CLL clones.45

(A) Application of mathematical modeling to data of patients with CLL in the setting of watch and wait. (B) Logistic, indeterminate, and exponential growth patterns discernible in CLL progression. Growth patterns are associated with marked differences in genetic composition (C), extent of clonal evolution, and growth dynamics (D and E). Reprinted from Gruber et al1 with permission.
(A) Application of mathematical modeling to data of patients with CLL in the setting of watch and wait. (B) Logistic, indeterminate, and exponential growth patterns discernible in CLL progression. Growth patterns are associated with marked differences in genetic composition (C), extent of clonal evolution, and growth dynamics (D and E). Reprinted from Gruber et al1 with permission.

(A) Clonal dynamics in the setting of therapeutic resistance. (B) WES of serial blood samples allows phylogenetic tree inferences, which highlight the presence of small subclonal populations driving relapse to ibrutinib therapy. (C) Clonal kinetics during ibrutinib treatment. Filled circles are measurements combining clonal fractions and absolute lymphocyte counts; empty circles are upper-bound estimates (1% of total CLL cells) for clones below the limit of detection; solid lines signify predicted kinetics for clones with 2 or more measurements; and dashed lines denote kinetics with minimal absolute growth rates for clones with only one measurement. (D) Droplet-based detection of resistance subclones at the time of treatment initiation. (E) Detection of RPS15-mutant specific single cells in pretreatment CLL samples as compared with a peripheral blood mononuclear cell control, using droplet-based single-cell detection. (F) Growth rates of relapsed CLL clones are several-fold higher than those of untreated CLLs.1,64,65 Reprinted from Burger et al64 with permission.
(A) Clonal dynamics in the setting of therapeutic resistance. (B) WES of serial blood samples allows phylogenetic tree inferences, which highlight the presence of small subclonal populations driving relapse to ibrutinib therapy. (C) Clonal kinetics during ibrutinib treatment. Filled circles are measurements combining clonal fractions and absolute lymphocyte counts; empty circles are upper-bound estimates (1% of total CLL cells) for clones below the limit of detection; solid lines signify predicted kinetics for clones with 2 or more measurements; and dashed lines denote kinetics with minimal absolute growth rates for clones with only one measurement. (D) Droplet-based detection of resistance subclones at the time of treatment initiation. (E) Detection of RPS15-mutant specific single cells in pretreatment CLL samples as compared with a peripheral blood mononuclear cell control, using droplet-based single-cell detection. (F) Growth rates of relapsed CLL clones are several-fold higher than those of untreated CLLs.1,64,65 Reprinted from Burger et al64 with permission.
Lineage tracing for the integrative analysis of clonal dynamics
Lineage-tracing methodologies, such as DNA barcoding, allow the high-resolution study of clonal representation and fitness in a polyclonal population (Figure 2C). Tools such as ClonTracer, GESTALT, and COLBERT, which leverage high-diversity DNA barcode libraries, have recently been introduced into primary cancer cells and cell lines to study interclonal dynamics, evolutionary trajectories, and mechanisms of drug resistance that encompass genetic features and their transcriptomic implications at lineage resolution.46-49 These are still new techniques, and it is anticipated that adaptation of such methodologies to CLL will help resolve the impact of CLL-associated genetic and epigenetic aberrations on cellular function.
Clonal dynamics in the setting of “watch and wait”
For over a decade, “watch and wait” (W/W) has been the mainstay approach for patients with CLL without symptomatic disease, although patients often progress to a point at which frontline treatment is warranted.50 In an effort to discover the genetic determinants of natural progression, several studies have used WES and/or genome-wide single-nucleotide polymorphism arrays to evaluate the mutational profiles of CLL. In a recent WES study, CLL samples were also collected across multiple time points during this pretreatment period (ranging from 2 to 5 samples per patient and covering 3-5 years).1 In aggregate, the analysis of clonal architecture of these samples has revealed that clonal equilibrium is very common during this period.2,26,51 Indeed, genetic alterations initially identified in a tumor at the time of diagnosis have been shown to remain stable over time, rather than the tumors acquiring new lesions. Hence, in the absence of a strong selective pressure, clonal stability appears to be the predominate pattern. However, Smith et al have reported that the methylation profiles of W/W patients yielded recurrent epigenetic changes primarily involving memory B-cell–specific polycomb repression complex 2 targets involved in chromatin remodeling and regulation of gene expression.51
In the most comprehensive study of the W/W period to date, Gruber et al evaluated growth patterns in CLL in the setting of natural disease progression (Figure 3A).1 They described distinct patterns of CLL growth in >100 patients with CLL and observed that even in the absence of therapy, patient tumors could be categorized on the basis of their CLL growth patterns. These patterns included logistic growth, which is sigmoidal and stabilizes at a certain steady-state level, and exponential, or unbounded, growth, as defined by <1000 × 109 cells/L and >1000 × 109 cells/L, respectively (Figure 3B). In an analysis of serial samples collected between diagnosis and first treatment from >100 patients with CLL, they found that CLLs exhibiting exponential growth tended to undergo clonal evolution, genetic complexity (ie, a larger number of CLL drivers, including tri[12] and unmutated IgH), and faster disease progression. Logistic growth tended to be more defined by clonal equilibrium, a narrower spectrum of genetic lesions (including the less aggressive del[13q] and mutated IgH), and a more indolent disease course (Figure 3C). Subclonal kinetics did not always match the growth patterns of the overall tumor; for example, CLLs characterized by logistic growth globally could nonetheless harbor subclones with declining, plateauing, or even exponential growth rates. In general, subclones harboring well-established CLL drivers (ie, TP53, ATM, XPO1, KRAS) had higher growth rates than parental or sibling subclones lacking said drivers, providing direct in vivo evidence of a selective growth advantage conferred by putative driver mutations (Figures 3D and 3E). A thorough understanding of the growth kinetics driven by a combination of genetic and/or epigenetic alterations is anticipated to be of great benefit to CLL prognostication and therapeutic decision making.
Clonal dynamics in therapeutic resistance
Although the frontline therapy of fludarabine, cyclophosphamide, and rituximab has long been the standard of care for younger patients with low-risk disease, a number of novel targeted therapies have been added to the CLL therapeutic landscape over the past 5 years and are rapidly gaining prominence in the first-line setting. These most prominently include ibrutinib (a BTK inhibitor) and idelalisib (a phosphatidylinositol 3-kinase inhibitor), which function to abrogate B-cell receptor signaling, and venetoclax (a BCL2 inhibitor) for high-risk patients or those who relapse after frontline treatment. Although these therapies have been highly effective with deep responses, many tumors do recur after a period of initial response (Figure 4A) and display a range of evolutionary patterns with recurrence.7,52,53
Clonal dynamics in chemotherapy resistance
Resistance to chemotherapy appears to employ diverse evolutionary strategies, often selecting for preexisting TP53 mutant clones harboring high genomic complexity (ie, unmutated IgH)54,55 and resulting in marked clonal evolution over the course of relapse.4,56 In a study of 59 patients in which WES was performed on matched samples before first-line fludarabine therapy and upon relapse, large clonal shifts representing both linear and branched evolution were observed, with the relapse clone already detectable before treatment in 30% of cases.4 Biallelic inactivation of TP53 and ATM was common in relapse clones, as well as additional mutations (ie, IKZF3) thought to enhance fitness in the face of fludarabine therapy.56-58
Clonal dynamics in resistance to targeted agents
In general, aggregate data across cancers regarding resistance to targeted therapies have revealed the employment of specific mechanisms of resistance directly related to the pathways impacted by the specific targeted agent. These include inactivating mutations in the target gene, activation of critical signaling pathways parallel to or downstream of the target, overactivation of an unrelated prosurvival pathway, and/or histologic transformation.59 In the case of CLL, resistance to ibrutinib, for example, has been found to predominantly involve mutation of its direct target, BTK (C481S), or converging mutations in BTK’s immediate downstream partner PLCG2 (leading to autonomous B-cell receptor activity).60,61 Other lesions associated with ibrutinib resistance include del(8p), which encompasses the TNF-related apoptosis-inducing ligand (TRAIL) receptor and confers resistance to TRAIL-induced apoptosis; clonal gain-of-function mutation of CARD11, previously reported to confer resistance to ibrutinib in diffuse large B-cell lymphoma and thought to activate the NF-κB pathway; mutation of ITPKB, a central feedback inhibitor of the B-cell receptor signaling pathway; and mutation of previously described CLL driver genes SF3B1 and TP53.20 Furthermore, CLLs treated with ibrutinib have been reported to undergo histologic transformation (in this case, a small subclonal population harboring an ITPKB mutation), which can underlie primary refractory disease or early progression.7,9,62,63 Clonal dynamics are dictated not only by intrinsic molecular features but also by environmental factors. For example, Gaiti et al performed multiplexed single-cell reduced-representation bisulfite sequencing of serial CLL samples before and during ibrutinib-associated lymphocytosis and showed evidence of genetically and epigenetically divergent lineages, marked by distinct transcriptional profiles, that were preferentially expelled from the lymph node and thus differentially responsive to therapy.36
The breadth of diverging and converging evolutionary mechanisms that have been observed in CLL resistance to date point to a broader question: Is resistance driven by acquisition of de novo mutations, or are resistance mutations already present before treatment? Thus far, the selection of rare clones with preexisting rather than de novo genetic lesions over the course of ibrutinib therapy has been supported by mathematical modeling,40 as well as by sensitive experimental detection of small populations of cells in the pretreatment population harboring resistance mutations, as shown in a study by Burger et al.64 For example, WES performed on 5 serial peripheral blood CLL samples of one particular patient allowed the detection of a minor subclone harboring a del(8p) at pretreatment and a dominant clone at relapse that was a progeny of the del(8p)-positive subclone but also harbored additional putative driver mutations in EIF2A and RPS15 (Figures 4A and 4B). Analysis of this patient’s CLL growth kinetics (using absolute lymphocyte counts) demonstrates that the parental del(8p) clone (clone 3 in Figure 4B) was declining at the time of ibrutinib initiation, whereas its EIF2A- and RPS15-containing progeny (clones 4 and 5) were exhibiting elevated daily growth rates in comparison (Figure 4C). These findings were validated through single-cell droplet-based polymerase chain reaction detection, which confirmed the presence of a small cell population associated with the resistant subclone in pretreatment samples but not in peripheral blood mononuclear cells from normal adult donors (Figures 4D and 4E). In aggregate, these findings suggest that time to clinically detectable relapse is determined not only by the presence of resistance-conferring mutations within subclonal populations but also by the size and growth rate of the drug-resistant clone at the time of treatment initiation.64 Growth rates of relapsed clones grow faster than clones from untreated CLLs1,64,65 (Figure 4F).
Like ibrutinib, resistance to venetoclax is also a product of active selection. Of note, although point mutations in apoptosis-related genes (including venetoclax target BCL2 [G101V]) have recently been reported, relapse clones also appear to employ diverse alternative potential resistance mechanisms.66,67 Herling et al reported 8 patients with CLL characterized by WES and methylation profiling before and after relapse to venetoclax that demonstrated signs of accumulating genomic instability (copy number alterations or aneuploidy); recurrent mutations in BTG1, NOTCH1, and TP53; and infrequent alterations in BRAF, CD274 (PD-L1), NOTCH1, RB1, SF3B1, and TP53 (divergent evolution).8 Furthermore, mutations that impact energy stress sensing (protein kinase A/adenosine 5′-monophosphate–activated protein kinase) signaling pathways and regulators of mitochondrial metabolism have also been implicated in venetoclax resistance.67
Clinical and therapeutic implications
CLL’s ability to evolve and adapt to broad chemotherapy and targeted therapy is a major challenge to successful treatment and durability of response. In addition to the 2 established clinical staging systems, Rai and Binet, which rely on clinical presentation for prognostication, new prognostic scoring systems also include routine screening for chromosomal aberrations and the wide array of somatic mutations associated with poor treatment response, progression-free survival, and overall survival.68 However, even these prognostic scoring systems cannot yet account for the fact that individual cancers do not always share a uniform combination of genetic or epigenetic abnormalities. Furthermore, an accurate projection of cancer growth requires knowledge of the tumor’s subclonal composition and the respective growth rates and fitness dynamics of the subclones therein.
A different strategy for prognostication is more in line with the concept of personalized medicine: To identify a tumor’s potential to evolve, predict its likely evolutionary trajectories and formulate therapy accordingly. Therapies can involve numerous strategies, including but not limited to those described in the subsections below.
Targeting clonal vs subclonal lesions
The question of whether to target the trunk vs the branches of a tumor’s evolutionary phylogenetic tree has been debated and is likely dependent on (1) the subclonal composition of the tumor in question, (2) the complex relationship between different genetic lesions within a target subclonal population, (3) the degree of genetic or epigenetic heterogeneity present within that subclone, and (4) the availability of relevant targeted agents. Targeting truncal alterations could theoretically lead to complete extinction of all malignant cells, but this strategy is limited by the dependence of subclones on the truncal target; it is possible that presence of coexisting genetic alterations may override sensitivity to the targeted agent. For example, Burger et al described a patient with an SF3B1 (G742D) mutation before fludarabine, cyclophosphamide, and rituximab that, upon relapse, was replaced with a clone harboring biallelic inactivation of TP53, trisomy 12, and a new mutation in SF3B1 (K666T).62 Prior knowledge of the subclonal driver mutations that will be most expanded after frontline chemotherapy would allow the combination of standard intervention chemotherapy with a targeted agent toward that subclonal driver.
Evolutionary herding
A second approach involves promoting stable interclonal equilibrium to prevent development of aggressive or resistant phenotypes.69 To achieve this, one can do the following:
Target the genetic or epigenetic mechanisms by which clones diversify early in disease;
Specifically target lesions resulting in unstable phenotypes and preserve those with more stable phenotypes;
Introduce combination treatments composed of a “sensitizing” agent as well as a truncal mutation–targeted agent, such that known escape mechanisms are blocked off and cells are thus constrained to the pathways affected by the targeted agent; and
Implement a “debulking” protocol whereby a broad cytotoxic chemotherapy is applied in combination with a targeted agent, such as to drastically reduce the heterogeneity of a population and thus the likelihood of a rare population carrying a preexisting mutation that bypasses the targeted agent, an approach increasingly employed in clinical trials.
Harnessing antitumor immunity
CLL is highly adaptable and constantly undergoing evolution, such that leveraging an equally adaptable immune system to help mitigate its expansion may be highly beneficial. The immune system may help to maintain cancer subclones in a state of equilibrium whereby clonal expansions are attenuated by adaptive immunity. How tumor cells evade immune predation is a current area of intense interest and ongoing study, and leveraging a typically dysfunctional CLL immune system also has its challenges.70
Despite these potential avenues, we are still in the early stages of understanding the nuanced clonal dynamics underlying CLL pathology. Several obstacles limit implementation of such approaches in the clinic, including the still relatively high cost of NGS, its lower bounds of sensitivity (false-positive findings if [or alternatively, when] <5% of allelic frequency), and its limited availability in the community hospital setting. As such, the strategies proposed in this article are of an exploratory nature, and additional studies are needed to further ascertain their predictive capacity at larger scale. However, given rapidly decreasing costs of sequencing and constant advances in sequencing technologies, we anticipate these approaches will become increasingly feasible. As we further our understanding of the clonal dynamics involved in disease, we expect such findings will be considered when interpreting outcomes (progression-free survival or overall survival) during clinical trials. Finally, the therapeutic strategies described above are particularly suited for CLL therapy because they are contingent on the availability of sequential tumor samples for the identification and prediction of evolutionary patterns; such approaches will be harder to implement in most solid or fast-growing tumors.
Conclusion and future directions
Analysis of clinical cases
Mathematical modeling using serial WBC counts and WES at diagnosis and immediately before therapy revealed the following:
The patient in clinical case 1 experienced a growth rate per year of 37% that followed a logistic growth pattern before treatment. Upon WES analysis, 4 major subclones were identified, 2 of which share a subclonal del(13q) mutation. Each clone had a different respective growth pattern, such that clones sharing a del(13q) had more similar, logistic growth, whereas one subclone lacking any identifiable CLL driver exhibited exponential growth. The patient was monitored for 8 years before requiring therapy and was started on a fludarabine/rituximab regimen, to which he has had a complete response.
The patient in clinical case 2 experienced a growth rate per year of 64% that followed an exponential growth pattern before treatment. In WES analysis of the relapsed population, it became clear that a subclone with multiple driver mutations (XPO1, del[13q], del[15q]) expanded more rapidly than its parent (differential growth rate of 57%/y). The patient was monitored for 3 years before requiring therapy and was started on a fludarabine/rituximab regimen, to which he relapsed and required secondary treatment after 6 years.
As exemplified by these clinical cases, genetics play a central role in the clonal dynamics of tumor evolution. Mutation of IgH confers a more favorable prognosis and usually associates with logistic tumor growth and low genomic complexity. Unmutated IgH usually co-occurs with strong CLL drivers and rapid exponential growth, a shorter time to treatment, and poor response to therapy.1 Whereas the patient in clinical case 1 exhibited overall clonal equilibrium from the time of diagnosis to treatment, active clonal evolution was evident in the patient in clinical case 2, who experienced larger shifts in clonal fractions, with more proliferative clones having greater genomic complexity than their parent clone. However, the presence of a subclone in the patient in clinical case 1 that exhibits exponential growth despite global logistic growth emphasizes the breadth of clonal dynamics that is possible even in more stable disease.
Ongoing characterization of clonal dynamics in CLL has yielded a growing understanding that clone size, growth kinetics, and genetic characteristics all play a role in disease progression and resistance. Newer studies using single-cell methodologies are also beginning to suggest that epigenetics play a role in clonal dynamics, one that perhaps intersects and cooperates with that of genetics. Though in its early stages, elucidation of evolutionary mechanisms at the genetic and epigenetic levels is anticipated to improve predictions of tumor growth rates, inform future clinical trials, contribute to the evaluation of drug response, and help in the formulation of future therapy combinations or series of treatments.
Acknowledgments
C.G. is supported by the American Society of Hematology Minority Medical Student Award Program and the National Institutes of Health Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship F31 Award (1F31CA239443-01). C.J.W. is a scholar of the Leukemia and Lymphoma Society, whose work is supported by the National Cancer Institute (1R01CA155010-01A1, P01CA206978, U10CA180861).
The authors are grateful to I. Bozic, M. Gruber, O. Olive, and A. Al’Khafaji for helpful discussions.
Correspondence
Catherine J. Wu, Dana-Farber Cancer Institute, Dana Bldg, 44 Binney St, Boston, MA 02115; e-mail: cwu@partners.org.
