
Abstract
Liver disease is an important cause of morbidity and mortality in patients with sickle cell disease (SCD). Despite this, the natural history of liver disease is not well characterized and the evidence basis for specific therapeutic intervention is not robust. The spectrum of clinical liver disease encountered includes asymptomatic abnormalities of liver function; acute deteriorations in liver function, sometimes with a dramatic clinical phenotype; and decompensated chronic liver disease. In this paper, the pathophysiology and clinical presentation of patients with acute and chronic liver disease will be outlined. Advice will be given regarding initial assessment and investigation. The evidence for specific medical and surgical interventions will be reviewed, and management recommendations made for each specific clinical presentation. The potential role for liver transplantation will be considered in detail.
Learning Objectives
Gain an understanding of the wide variety of liver pathology and disease encountered in patients with SCD
Develop a logical approach to evaluate liver dysfunction and disease in patients with SCD
Review the evidence basis for specific medical and surgical interventions for acute and chronic liver complications
Clinical case
We report the case of a man of African-Caribbean descent, diagnosed with sickle cell disease (SCD; genotype HbSS) as a child. He had been followed-up in our institution from the age of 14 years. Past medical history included common complications of SCD: gallstone disease, priapism, and multiple admissions with acute pain crises. He did not tolerate hydroxyurea. He had no history of alcohol excess.
He developed a chronic abnormality of liver function at the age of 24 years. This was characterized by hyperbilirubinemia (predominantly conjugated) and a mixed abnormality of the liver enzymes. The patient reported scleral icterus and right upper quadrant abdominal pain during vaso-occlusive crises. Abdominal imaging at this time showed gallstones, hepatomegaly, and splenic infarction. A laparoscopic cholecystectomy at age 25 years did not improve symptoms.
At age 26 years, the patient presented to the emergency department with a further episode of jaundice and abdominal pain, in the context of a sickle crisis precipitated by viral infection. At clinical examination, he was found to be deeply icteric, febrile at 37.8°C, with right upper quadrant tenderness elicited on abdominal palpation. Liver function tests showed an acute deterioration: bilirubin, 34.4 mg/dL (predominantly unconjugated); alanine transaminase, 341 IU/L; alkaline phosphatase, 430 IU/L; albumin, 32 g/L. Renal function and clotting were normal. Cross-sectional imaging demonstrated hepatomegaly, mild intrahepatic bile duct dilation without evidence for bile duct stones, and patency of hepatic vasculature. hemoglobin S (HbS) fraction was 80%. The patient was managed as having an acute sickle liver in the context of an acute vaso-occlusive crisis. Treatment included IV fluids, antibiotics, analgesia, and exchange blood transfusion (EBT) with the aim of reducing the HbS fraction to <30% to 40%. With this regimen, symptoms and acute liver dysfunction resolved, but bilirubin did not return to the preepisode baseline.
Over the next 4 years, the clinical phenotype was of a progressive predominantly cholestatic abnormality of liver function, punctuated by episodes of acute liver dysfunction similar to that described in the previous paragraphs during sickle crises (Figure 1). Further liver investigations performed included abdominal computed tomography (CT) and liver magnetic resonance imaging (MRI), which demonstrated (Figure 2) hepatomegaly with an irregular liver outline suggesting the development of chronic fibrotic disease, and right and left intrahepatic duct dilatation, suggestive of a cholangiopathy. Laboratory results revealed negative viral serology (for hepatitis B, hepatitis C, and hepatitis E), and a negative liver autoantibody screen (excluding liver autoimmune disease). Ferritin levels in steady state were markedly raised at 3000 to 4000 µg/L. An R2-MRI of the liver showed a liver iron concentration of 7.9 mg Fe/g dry weight consistent with transfusion-related iron overload.
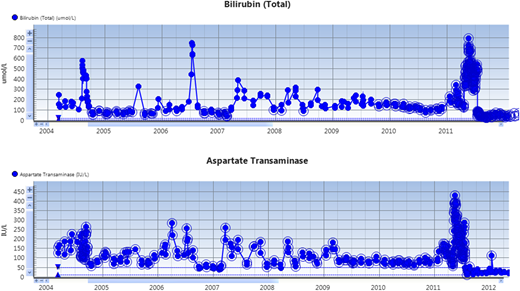
Serial liver function tests in the patient. Note the acute increases in serum bilirubin and aspartate aminotransferase (AST) during vaso-occlusive crises. Note: the units for bilirubin on the chart are SI (µmol/L).
Serial liver function tests in the patient. Note the acute increases in serum bilirubin and aspartate aminotransferase (AST) during vaso-occlusive crises. Note: the units for bilirubin on the chart are SI (µmol/L).
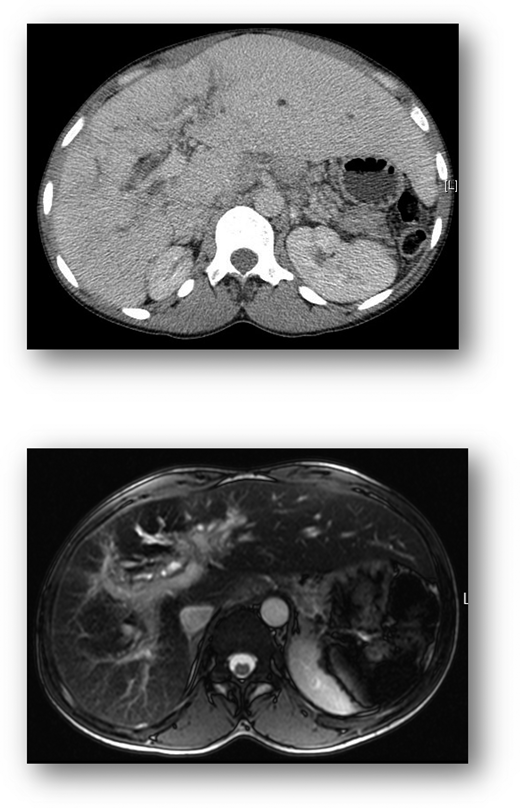
Cross-sectional imaging. CT liver (top) demonstrates hepatomegaly and irregular liver outline suggesting established chronic liver disease. MRI of the liver (bottom) shows diffuse injury of the intrahepatic bile ducts, indicative of an established cholangiopathy
Cross-sectional imaging. CT liver (top) demonstrates hepatomegaly and irregular liver outline suggesting established chronic liver disease. MRI of the liver (bottom) shows diffuse injury of the intrahepatic bile ducts, indicative of an established cholangiopathy
Regular exchange blood transfusions were instituted with an HbS percentage target of <30% to 40%. Iron chelation was initiated with deferasirox. Ursodeoxycholic acid was commenced. Despite these measures, the deterioration in liver function was progressive. Hydroxyurea was instituted in place of exchange transfusion for a period of 6 months, without improvement from the perspective of liver dysfunction. By 2011, the patient had developed a clinical picture of decompensated chronic liver disease. Decompensation was manifest by the development of ascites, as well as deterioration in synthetic function (reduction in serum albumin to <30 g/L). He underwent formal hepatological assessment for liver transplantation. The projected mortality from his liver disease (>20% at 1 year) justified assessment at this time. A rigorous evaluation excluded significant sickle-related heart, lung, kidney, or brain dysfunction. A protocol was developed with the sickle hematology team for optimal hematologic management in the peri- and postoperative period. This included EBT to maintain the HbS percentage at <30 to 40.
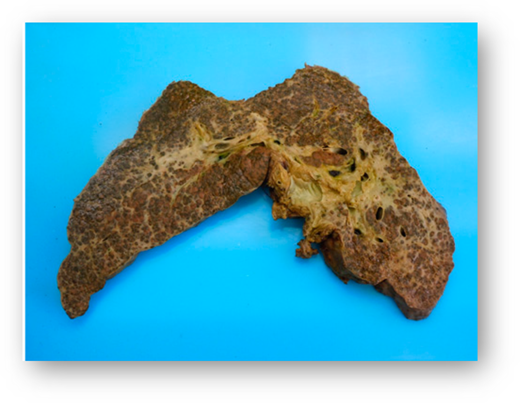
He underwent liver transplantation on 15 July 2011. Immunosuppression was as per our unit’s protocol with steroids and tacrolimus. The immediate postoperative recovery was good. The explanted liver was nodular and weighed 3700 g (Figure 3); histology demonstrated sickle cells in sinusoids, and a biliary pattern of injury with a secondary sclerosing cholangitis; also seen were moderate siderosis and a biliary abscess. Liver enzymes normalized within 1 month posttransplant while he was maintained on an EBT program. Seven years post liver transplantation, his general health is excellent as is liver graft function. Sickle-related complications included: 2 admissions with sepsis and proteinuria treated with candesartan. Regular EBT continues. There have been no significant issues related to the liver graft.
Introduction
In developed countries, SCD is a debilitating chronic disorder with significant morbidity due to end-organ damage. The liver is one of the affected organs, resulting in “sickle hepatopathy.” Some end-organ damage in SCD has a well-recognized natural history with evidence-based treatment algorithms: this is the case with renal disease, cardiopulmonary disease, and cerebrovascular disease (in children). However, the natural history and pathogenesis of liver disease is not well characterized, and the evidence basis for interventions is not robust, despite this end-organ damage being an important cause for morbidity and mortality.1,2
What causes liver disease in patients with sickle cell disease?
“Sickle hepatopathy” is an umbrella term, encompassing the range of liver disease encountered in patients with sickle cell anemia. As such, it includes diverse hepatic pathology arising from a variety of insults to the liver that can occur in these patients. It occurs predominantly in patients with homozygous HbSS disease, and to a lesser extent in patients with hemoglobin SC disease or HbS β-thalassemia.3 Liver disease can result from:
the effects of sickling of erythrocytes within the vasculature of the liver, with consequent hypoxic liver injury, particularly affecting the centrilobular region and biliary system;
complications related to the multiple blood transfusions some patients require, including viral hepatitis and iron overload;
gallstones, causing gallbladder pathology and biliary obstruction; or
coincidental liver pathology (eg, a young female patient with autoimmune liver disease).4
Classification of sickle hepatopathy
| Acute liver disease | Chronic liver disease | |
|---|---|---|
| Related to sickling process | • Acute sickle hepatic crisis • Hepatic sequestration • Sickle intrahepatic cholestasis | • Chronic cholestasis • Biliary-type cirrhosis |
| Related to multiple blood transfusion | • Viral hepatitis B and C | • Chronic viral hepatitis • Iron overload |
| Miscellaneous | • Cholelithiasis • Budd-Chiari syndrome • Hepatic abscess/biloma | • Cholelithiasis • Coincidental chronic liver disease, eg, autoimmune |
| Acute liver disease | Chronic liver disease | |
|---|---|---|
| Related to sickling process | • Acute sickle hepatic crisis • Hepatic sequestration • Sickle intrahepatic cholestasis | • Chronic cholestasis • Biliary-type cirrhosis |
| Related to multiple blood transfusion | • Viral hepatitis B and C | • Chronic viral hepatitis • Iron overload |
| Miscellaneous | • Cholelithiasis • Budd-Chiari syndrome • Hepatic abscess/biloma | • Cholelithiasis • Coincidental chronic liver disease, eg, autoimmune |
Reproduced from Tavabie and Suddle5 with permission.
How common is liver disease in SCD?
The incidence of sickle hepatopathy is difficult to define. Abnormalities in standard liver function tests are common in sickle cell anemia, and do not necessarily reflect intrinsic liver disease. For example, a moderate increase in bilirubin (predominantly unconjugated) and aspartate transaminase may be a consequence of hemolysis. Prevalence of liver dysfunction in SCD has previously been reported as 10%.6 The prevalence of cirrhosis in autopsy series is up to 30%, indicating that chronic liver disease is an important consideration in this group of patients.7,8
How should potential liver disease be investigated?
As outlined, assessment of standard liver function tests at baseline or steady state can be difficult, unless very normal or very abnormal. Minor abnormalities may reflect hemolysis more than intrinsic liver injury. More significant derangements are encountered in the acute liver syndromes, as described in the following paragraphs. In the setting of chronic liver disease, a progressive increase in the conjugated fraction of total bilirubin and reduction in serum albumin are ominous signs. All patients should have a screen to identify liver disease that has resulted from transfusion of blood or is coexistent; this includes hepatitis viral serology, a liver autoantibody screen (including antinuclear antibodies, anti–smooth muscle antibodies, and anti–liver kidney microsomal antibodies), ferritin, and copper studies. Ferritin is limited as a marker of iron burden in SCD because it can be affected by both liver disease and inflammation. Instead, magnetic resonance imaging is recommended to detect liver iron load such as R2-MRI or T2* measurements.9
Appropriate imaging studies include abdominal ultrasound, cross-sectional imaging with CT or MRI scan, and indirect cholangiography. The liver is usually enlarged and splenic infarction characteristic. Irregularity of the liver outline is suggestive of established fibrotic disease. Magnetic resonance cholangiography will identify cholelithiasis and choledocholithiasis as well as cholangiopathic change.10
The role of liver biopsy
This is an important clinical question. The histologic changes occurring in acute and chronic liver disease in patients with SCD have been characterized: sinusoidal obstruction due to sickling with variable centrilobular and hepatocyte damage in the acute syndromes, and cholangiopathy, biliary type cirrhosis, and iron overload in the chronic setting.4,7,11 However, percutaneous liver biopsy can be associated with increased risk of complications in SCD, principally hemorrhage. In a series from our center, 36% of patients undergoing liver biopsy developed severe bleeding complications and 28% died.12 The majority of patients who developed complications were in acute sickle crisis at the time of the biopsy. Importantly, the authors did not feel that histology significantly changed the management of these patients.
In general, assessment of the severity of liver disease and likely predominant etiology(ies) can be made after thorough clinical assessment and appropriate laboratory and radiological investigation. Liver biopsy is relatively contraindicated, especially in the acute liver syndromes. I would recommend that it only be considered if results will materially affect management, in conjunction with specialist hepatology input, and via the trans-jugular route to minimize the risks of bleeding. An example of when biopsy may be appropriate is when diagnosing autoimmune hepatitis in a young patient with chronic liver disease and suggestive serology.10,13 The role of noninvasive investigations for liver fibrosis requires further validation in this group of patients.14
Acute liver disease
Acute sickle liver
The acute episode of liver dysfunction outlined in the case presentation is best characterized as acute sickle liver. This occurs in ∼10% of patients with SCD, usually in the context of a vaso-occlusive crisis. The clinical presentation is commonly right upper quadrant pain, jaundice, fever, and tender hepatomegaly. The serum bilirubin is usually <15 mg/dL, and transaminases are usually in the hundreds (as opposed to thousands). Renal function is preserved, and the prothrombin time is normal. Management is full supportive care and may include EBT with the aim of reducing the HBS% to <30% to 40%. Full reversal with these measures is the usual clinical course.15
Acute sickle intrahepatic cholestasis
The most severe clinical phenotype of sickle hepatopathy is acute sickle intrahepatic cholestasis, which is often fatal.16 It is uncommon. The pathophysiology is widespread sickling within the sinusoids, with resultant ischemia and hepatocyte injury.2 The clinical presentation can be similar to acute sickle liver with right upper quadrant pain and fever, but with marked jaundice, renal failure, and bleeding diathesis. Striking jaundice can develop (levels of up to 270 mg/dL are reported); there is marked elevation in transaminases (can be in the thousands), in association with renal failure and coagulopathy.17 Full supportive management is instituted, and transfer to an intensive care unit at an early stage is recommended. There is some evidence for the use of exchange transfusion to maintain the HbS percentage at <30%.18,19
Hepatic sequestration
The patho-physiology of hepatic sequestration is liver enlargement caused by intrahepatic trapping of red cells, accompanied by acute anemia. Patients often present with right hypochondrial pain accompanied by an enlarging, tense liver. The laboratory features are acute anemia, reticulocytosis, mild to moderate hyperbilirubinemia, and relatively normal liver enzyme levels. Management is supportive: transfusion may be indicated for symptomatic anemia.20
Acute gallstone disease
Gallstones are a common occurrence in SCD, present in up to 30% of children and 70% of adults.21 Symptomatic biliary tract disease occurs in ∼20% of patients.22 The management of acute cholecystitis, ascending cholangitis, and gallbladder empyema is not different from the general population and consists of broad-spectrum antibiotics that should include cover for Salmonella species and anaerobic organisms. Elective cholecystectomy is appropriate after acute complications such as cholecystitis or pancreatitis settle. The role of elective cholecystectomy for asymptomatic cholelithiasis remains controversial.23
Management recommendations for acute liver disease in SCD are summarized in Table 2.
Management recommendations in acute sickle hepatopathy
| Clinical scenario | Clinical/investigative data | Management recommendations |
|---|---|---|
| Gallstones | May be asymptomatic Right upper quadrant pain Cholangitis | Cholecystectomy for symptomatic gallstones ERCP with duct clearance for bile duct stones |
| Acute sickle hepatic crisis | Vaso-occlusive crisis RUQ pain/jaundice/leukocytosis Bilirubin <15 mg/dL ALT rarely >300 IU/L | Supportive Consider exchange blood transfusion |
| Sickle cell intrahepatic cholestasis | Vaso-occlusive crisis RUQ pain, leukocytosis, fever, striking jaundice Very high bilirubin ALT can be in 1000s Coagulopathy Renal failure | Full supportive management Exchange blood transfusion |
| Hepatic sequestration | Enlarging liver RUQ pain Anemia Reticulocytosis | Supportive Transfusion |
| Clinical scenario | Clinical/investigative data | Management recommendations |
|---|---|---|
| Gallstones | May be asymptomatic Right upper quadrant pain Cholangitis | Cholecystectomy for symptomatic gallstones ERCP with duct clearance for bile duct stones |
| Acute sickle hepatic crisis | Vaso-occlusive crisis RUQ pain/jaundice/leukocytosis Bilirubin <15 mg/dL ALT rarely >300 IU/L | Supportive Consider exchange blood transfusion |
| Sickle cell intrahepatic cholestasis | Vaso-occlusive crisis RUQ pain, leukocytosis, fever, striking jaundice Very high bilirubin ALT can be in 1000s Coagulopathy Renal failure | Full supportive management Exchange blood transfusion |
| Hepatic sequestration | Enlarging liver RUQ pain Anemia Reticulocytosis | Supportive Transfusion |
Adapted from Tavabie and Suddle5 with permission.
ALT, alanine aminotransferase; RUQ, right upper quadrant.
Chronic liver disease
The patient in the case history developed progressive chronic liver disease, culminating in decompensated cirrhosis. The natural history of this chronic phase of sickle hepatopathy is not well defined. A particular challenge is identifying which patients are at risk of developing progressive liver damage secondary to recurrent intrahepatic sickling. Patients with chronic cholestasis often report noting increased icterus during previous acute crises. The evidence basis for medical treatments is not strong. There is a need for good-quality, prospective studies to define natural history and provide evidence for specific interventions. The prevalence of cirrhosis in autopsy studies has been between 16% and 29%,8 indicating that chronic liver disease is a real and important clinical issue for patients with SCD. Pathology encountered in those with chronic cholestasis includes a secondary sclerosing cholangitis and biliary-type cirrhosis.4,24
An important question that remains unanswered is whether any specific intervention in the patient with established chronic liver disease will modulate the risk of progression to end-stage liver disease. There is evidence from case histories supporting the use of regular EBT to maintain the HbS percentage at <30% to 40%.25,26 The use of ursodeoxycholic acid, a drug commonly prescribed in cholestatic liver disease, can be considered. It may be effective, and will likely not be toxic. Hydroxyurea may be considered if the patient has frequent vaso-occlusive crises. The registration studies defining the use of hydroxyurea for patients with SCD showed that those treated with the drug had lower total bilirubin levels than matched placebo controls; whether this correlates with less severe liver disease (rather than less hemolysis) is unclear.27 There are no data regarding the combination of hydroxyurea and EBT to prevent liver disease progression.28
Iron overload and chronic viral hepatitis
Iron chelation for iron overload is recommended.29 The novel directly acting antiviral therapies have changed the treatment landscape in hepatitis C virus treatment demonstrating very good safety profile and excellent viral eradication rates (>95%). The vast majority of patients can now be treated with a ribavirin-free drug regimen.30,31 Treatment of chronic hepatitis B should be with the potent nucleot(s)ide analogs, which very effectively suppress viral replication.32
What is the role for liver transplantation?
The case presentation outlined the history of a patient who suffered with acute sickle liver syndromes and progressive chronic liver disease despite interventions such as regular EBT. Is there an evidence basis for recommending liver transplantation for patients with a similar clinical phenotype?
Worldwide experience of liver transplantation as a treatment of end-stage liver disease in patients with SCD is slowly accruing. Experience is currently limited, and several questions remain unanswered. The poor results reported in early case series, with crude mortality rates of ∼60% at 1 year posttransplant, have improved in later studies. Better results are likely a consequence of improved patient selection and perioperative management of SCD.33,34
What conclusions can be drawn from the current data? There appear to be 2 clinical phenotypes that benefit most from liver transplantation. The first is the patient with end-stage liver disease, but without significant sickle-related complications of the heart, lungs, kidneys, and brain. The second group is those with liver disease coincident to SCD; for example, those with autoimmune-related chronic liver disease.1,10,35,36 Regarding potential contraindications, transplantation in those with the severe acute hepatic dysfunction has been associated with very poor outcomes.37
Recommendations for perioperative management of SCD include regular EBT to maintain HbS percentage at <30% to 40%; this is not evidence based and is largely extrapolated from studies involving nontransplant surgery. Recurrent sickle-related damage in the liver graft is reported.38,39 Most groups advocate maintenance of exchange transfusion posttransplant to prevent this complication.
Management recommendations for chronic liver disease are summarized in Table 3.
Management recommendations for chronic sickle hepatopathy
| Clinical scenario | Clinical/investigative data | Management recommendations |
|---|---|---|
| Chronic cholestatic liver disease | ? History of acute crises Chronic elevation in bilirubin Exclude coincidental liver disease | Longitudinal assessment Consider regular exchange transfusion Consider ursodeoxycholic acid |
| End-stage chronic liver disease | Often biliary type cirrhosis Sclerosing cholangitis type injury reported Exclude coincidental liver disease (eg, autoimmune hepatitis) | Management of complications of cirrhosis Role of liver transplant not defined Consider if • young patient • predominant end-organ damage liver |
| Iron overload | Elevated ferritin MRI for iron concentration | Chelation when liver iron concentration >7 mg Fe/g dry weight |
| Viral hepatitis | Chronic hepatitis B virus Chronic hepatitis C virus | Treatment as per normal guidelines ? Avoidance of regimens with ribavirin for hepatitis C virus |
| Gallstone disease | Diagnosis by standard imaging techniques | Cholecystectomy-prophylactic controversial ERCP and duct clearance |
| Clinical scenario | Clinical/investigative data | Management recommendations |
|---|---|---|
| Chronic cholestatic liver disease | ? History of acute crises Chronic elevation in bilirubin Exclude coincidental liver disease | Longitudinal assessment Consider regular exchange transfusion Consider ursodeoxycholic acid |
| End-stage chronic liver disease | Often biliary type cirrhosis Sclerosing cholangitis type injury reported Exclude coincidental liver disease (eg, autoimmune hepatitis) | Management of complications of cirrhosis Role of liver transplant not defined Consider if • young patient • predominant end-organ damage liver |
| Iron overload | Elevated ferritin MRI for iron concentration | Chelation when liver iron concentration >7 mg Fe/g dry weight |
| Viral hepatitis | Chronic hepatitis B virus Chronic hepatitis C virus | Treatment as per normal guidelines ? Avoidance of regimens with ribavirin for hepatitis C virus |
| Gallstone disease | Diagnosis by standard imaging techniques | Cholecystectomy-prophylactic controversial ERCP and duct clearance |
Adapted from Tavabie and Suddle5 with permission.
ERCP, endoscopic retrograde cholangiopancreatography.
Summary
Liver disease in patients with SCD arises as a consequence of a wide variety of insults to the liver that can occur during the lifetime of patients and results in a diverse range of hepatic pathology. The term sickle hepatopathy encompasses this diverse pathology. The clinical phenotype of liver disease can vary from asymptomatic to acute and life threatening. The natural history of sickle hepatopathy is not well defined or characterized. Assessment of patients requires attention to detail, and a thorough evaluation of clinical, laboratory, and radiologic data. Liver biopsy is associated with a high risk of complications, and should only be necessary in the minority. The evidence basis for therapeutic interventions is not robust. EBT is widely used in the acute liver syndromes and in chronic liver disease. Liver transplantation may have a role in highly selected patients with decompensated chronic liver disease.
Correspondence
Abid R. Suddle, Institute of Liver Studies, King’s College Hospital, London SE5 9RS, United Kingdom; e-mail: abid.suddle@nhs.net.
