
Abstract
Osteonecrosis, a form of ischemic bone injury that leads to degenerative joint disease, affects ∼30% of people with sickle cell disease. Although osteonecrosis most commonly affects the femoral head (often bilaterally, with asymmetric clinical and radiographic progression), many people with sickle cell disease also present with multifocal joint involvement. We present the case of a young woman with bilateral osteonecrosis of the femoral head at varying stages of progression; we also highlight other important comorbid complications (eg, chronic pain requiring long-term opioids, debility, and social isolation) and postoperative outcomes. In this review, partly based on recommendations on osteonecrosis management from the 2014 evidence-based report on sickle cell disease from the National Heart, Lung and Blood Institutes, we also discuss early signs or symptoms of osteonecrosis of the femoral head, radiographic diagnosis and staging criteria, hydroxyurea effect on progression to femoral head collapse, and surgical outcomes of total hip arthroplasty in the modern era. In summary, we failed to find an association between hydroxyurea use and femoral head osteonecrosis; we also showed that evidence-based perioperative sickle cell disease management resulted in superior postoperative outcomes after cementless total hip arthroplasty in sickle cell–related osteonecrosis of the femoral head.
Learning Objectives
Understand that recurrent vaso-occlusive episodes and acute chest syndrome, not hydroxyurea use, predispose to osteonecrosis in sickle cell disease
Understand that perioperative management of sickle cell disease improves outcomes for people undergoing total hip arthroplasty with cementless prostheses
Case study
A 19-year-old woman with sickle cell anemia (hemoglobin [Hb] SS genotype) presents with left hip pain. Her past medical history is notable for frequent painful vaso-occlusive episodes (VOEs), recurrent acute chest syndrome (ACS), osteonecrosis of the right femoral head (R-ONFH), abnormal transcranial Doppler (TCD) findings, asthma, low bone mineral density, and vitamin D deficiency. As of December 2018, she had been hospitalized 6 times for VOEs and recurrent abdominal pain in the past 12 months. She was eventually diagnosed with gastric ulcers and choledocholithiasis in March 2018. She reports 10 lifetime episodes of ACS, with the most recent occurring in 2017. Although she never required emergent red blood cell (RBC) exchange transfusions for ACS treatment, she often received simple RBC transfusions during her hospitalizations. She was last transfused in March 2018 before she underwent an endoscopic retrograde cholangiopancreatography to remove the common bile duct stone felt to be causing her recurrent abdominal pain. She had previously undergone an uncomplicated tonsilloadenoidectomy for sleep apnea.
Although she had no history of overt stroke, TCD studies—obtained in childhood—showed stable narrowing of the supraclinoid segments of the right and left internal carotid arteries. She underwent chronic RBC transfusions from 2005 to 2009 and later, transitioned to hydroxyurea (HU) after her family declined ongoing transfusions. Given her inconsistent adherence to HU, the medication was discontinued in 2015. She developed progressive transfusional hepatic iron overload with the following liver iron content (in milligrams per gram dry weight) on T2* magnetic resonance imaging (MRI): June 2010 9.0 → July 2013 9.0 → July 2015 7.2 → November 2017 2.8.
She initially presented with right hip pain in early 2011 (age 12 years old); a plain radiograph of the right hip obtained in February 2011 was reportedly normal. Plain pelvic films from January 2013 (age 14 years old) showed slight flattening of her right femoral head and subtle sclerosis in her left hip. She was diagnosed with R-ONFH (Steinberg stage III) and referred for physical therapy and rehabilitation, which began in February 2013. Because she was instructed to minimize weight bearing, she mobilized on a wheelchair and ambulated on crutches for short distances. Follow-up plain hip films from April 2013 showed progressive flattening of her right femoral head (Steinberg stage IV) and normal appearance of her left hip; thus, her R-ONFH progressed within 3 months despite physical therapy and decreased weight bearing. In September 2013, bilateral hip MRI showed mild flattening of her right femoral head, necrosis, and a small effusion in her right hip joint space; she had a small area of osteonecrosis (OST) on the left femoral head without flattening. She began homeschooling because of persistent hip pain, which she rated a 4 to 7 of 10 on the visual analog pain scale.
She was evaluated by orthopedic surgery in February 2014 (age 15 years old), and repeat plain films of her hips showed R-ONFH progression with reduced joint space, sclerosis, and cysts (Steinberg stage V); the left femoral head had minimal flattening (Steinberg stage II). She continued with physical therapy and minimal weight bearing. In January 2015 (age 16 years old), orthopedics recommended right total hip arthroplasty (THA) for intractable pain, but her family declined surgical intervention. She initiated immediate release morphine 30 mg by mouth twice daily for her chronic hip pain. In July 2015, dual X-ray absorptiometry (DXA) scans of her left hip and lumber spine showed areal bone mineral density-for-age Z scores of −2.7 and −3.4, respectively. She was started on calcium and vitamin D supplementation. She eventually underwent a right THA and left hip core decompression in October 2016 (age 17 years old). Her hip pain and mobility significantly improved postoperatively; she enrolled in college and began to wean off morphine by December 2016.
In July 2017 (age 18 years old), she reinitiated HU because of ongoing VOEs and ACS. Her most recent laboratory values showed improved HU adherence with Hb 9.2 g/dL, fetal hemoglobin (Hb F) 22.9%, mean corpuscular volume (MCV) 109, platelets 323 000, absolute neutrophil count 5090, and absolute reticulocyte count 257 000. She endorsed occasional left hip discomfort with overuse but denied daily debilitating hip pain. Repeat DXA scans from November 2017 showed improved areal bone mineral density-for-age Z scores of −1.5 in her left hip and −2.7 in her lumbar spine. In April 2018, plain pelvic films showed a prosthetic right hip and left hip sclerosis without flattening (Steinberg stage II). Figure 1 shows bilateral osteonecrosis of the femoral head (ONFH) progression on serial plain pelvic films from April 2013 to April 2018. Her occasional left hip pain was alleviated by intermittent doses of oral hydromorphone. Pelvic MRI from September 2018 showed stable right hip prosthesis and progressive OST of the left femoral head with slight fragmentation and flattening (stage III). She was referred back to orthopedics in December 2018 for consideration of a left THA.
![Serial plain films of the pelvis showing ONFH progression from (A) April 2013 (flattening of right femoral head [Steinberg stage IV]; normal on left), (B) February 2014 (reduced joint space; sclerosis and cysts on the right [Steinberg stage V] and minimal collapse without flattening on the left [Steinberg stage II]), and (C) April 2018 (prosthetic right hip and sclerosis without flattening of the left femoral head [Steinberg stage II]).](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2019/1/10.1182_hematology.2019000038/5/m_hem2019000038cf1.png?Expires=1767767154&Signature=NhF8GaOv95ts5RCC8JBMusEnLja3GTcM0vHDWpBJQRxfGkl2L-fNh2hdSPWO~Cnysos2RB3KrTJH7U8C~fXqyVyJG~Nwtmf7fVXRl~1ECnY19nTL1y6zgQRdm83Ne~yznda0ACQLpjgWicIerzxZhK52RmzoPv~EZ06F9wZYjpSmKRcRg9~1pZJpnin-OOyb0RfxxlmBJ3jokRpQQ~3nTxiT~6STl5ge-RlcmfL6cYwqrBnjo~vfpGR1dMczbInHdCBpY6lGuCagUNQY3HULkyoBreY1Llkve9mczHsyfsVBaxJpLmaSlKDIIuSMD2G9uZSvmSuvqFKYMu2V0cTX2A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Serial plain films of the pelvis showing ONFH progression from (A) April 2013 (flattening of right femoral head [Steinberg stage IV]; normal on left), (B) February 2014 (reduced joint space; sclerosis and cysts on the right [Steinberg stage V] and minimal collapse without flattening on the left [Steinberg stage II]), and (C) April 2018 (prosthetic right hip and sclerosis without flattening of the left femoral head [Steinberg stage II]).
Serial plain films of the pelvis showing ONFH progression from (A) April 2013 (flattening of right femoral head [Steinberg stage IV]; normal on left), (B) February 2014 (reduced joint space; sclerosis and cysts on the right [Steinberg stage V] and minimal collapse without flattening on the left [Steinberg stage II]), and (C) April 2018 (prosthetic right hip and sclerosis without flattening of the left femoral head [Steinberg stage II]).
OST in sickle cell disease
Sickle cell disease (SCD) primarily manifests as a vascular disease where deformed and rigid RBCs aggregate and adhere to the vascular endothelium. Dense polymers of sickled RBCs, as well as activated leukocytes and platelets, occlude the microvasculature and obstruct downstream perfusion of tissues and organs. The ensuing vaso-occlusion causes recurrent, unpredictable, and excruciatingly painful episodes; hemolytic anemia; inflammation; and end organ damage. Osteonecrosis (OST) (also known as avascular, aseptic, or ischemic necrosis) is a prevalent skeletal complication of SCD attributed to repeated ischemia-reperfusion injury of susceptible articular surfaces.1,2 Recurrent painful VOEs, the hallmark of SCD, compromise blood flow to all joints and increase the likelihood of multifocal OST. Because the femoral head lacks collateral blood flow, it is particularly vulnerable to microvascular insults. ONFH typically affects both hips and often coexists with OST in the shoulders, knees, and other joints.3-5
SCD is the most common cause of pediatric ONFH; an estimated 50% of adults with homozygous SCD (Hb SS) will develop ONFH by age 35 years old.6 Systemic steroids, alcohol use, smoking, thrombophilia, cardiovascular disease, hyperlipidemia, and joint trauma increase the risk of ONFH in the general population. For people with SCD, risk factors for ONFH include older age, male gender, high body mass index, leukopenia, Hb SS with concurrent α-thalassemia trait (with relatively high Hb), frequent VOEs, and history of ACS.7,8 Although our patient did not have concomitant α-thalassemia trait, she clearly had recurrent VOEs and multiple episodes of ACS. Her bilateral ONFH progressed at different rates, possibly because of preferential weight bearing. Although her clinical symptoms seemed to match the imaging studies of progressive hip joint involvement with OST, clinicoradiographic correlation does not reliably occur in clinical practice.
We recently showed an independent association between ONFH and low bone mineral density in children and adolescents with SCD.9 ONFH can also predispose to chronic SCD pain,9,10 opioid dependence, and high utilization of acute medical care11 ; thus, ONFH negatively impacts health-related quality of life for people with SCD and further escalates already exorbitant health care costs.12,13 Our case also illustrates how low bone mineral density can coexist with ONFH. Although calcium and vitamin D supplementation aided our patient’s bone mineral density recovery, her improved mobility after right THA also played a key role, because mechanical loading from muscle mass and physical activity stimulate bone accrual and promote skeletal strength.14
ONFH prevalence in the modern era
In collaboration with researchers from the Center for Oncology Hematology Outcomes Research and Training at the University of California, Davis, we identified >6200 people with SCD across all nonfederal hospitals and emergency departments in California using hospital discharge data from California’s Office of Statewide Health Planning and Development (OSHPD; 1991-2013).8 After adjusting for birth cohort and accounting for competing risk of death, we found a 22% cumulative incidence rate for ONFH. Investigators from the Cooperative Study of Sickle Cell Disease (CSSCD; 1979-1981), a natural history cohort study, previously investigated ONFH prevalence and risk factors in >3200 people with SCD (ages 5-66 years old) recruited from 23 outpatient centers across the United States. In a subanalysis of almost 2600 CSSCD subjects, Milner et al6 found a 10% prevalence of ONFH using plain hip films at study entry. They prospectively monitored >1700 people without ONFH at baseline for 3 years and reported a 9% cumulative incidence of ONFH confirmed by plain radiographs. When they stratified by SCD genotype, they found ONFH incidence rates (per 100 patient-years) of 4.47 in Hb SS with α-thalassemia trait, 2.35 in Hb SS alone, and 1.91 in Hb SC (compound heterozygote Hb S and C).
The CSSCD strengths include its prospective study design and ONFH correlation with SCD genotypes. Solely using plain radiographs to detect ONFH may have accounted for their lower cumulative ONFH incidence compared with our OSHPD cohort study that included MRI for early-stage ONFH detection. The OSHPD dataset lacks reliable laboratory data, and therefore, we could not stratify by SCD genotype. We found an overall ONFH incidence rate ratio of 2.83 per 100 person-years, which was comparable with that of the Hb SS subset in the study by Milner et al.6 Our study was also susceptible to selection bias, because the OSHPD comprises people with SCD presenting to emergency rooms and hospitals (ie, those with a more severe phenotype) compared with the CSSCD subjects, who were enrolled from ambulatory clinics. Because the OSHPD dataset lacks reliable radiographic data, we used International Classification of Disease, 9th revision, Clinical Modification diagnosis codes only, and therefore, we may have inadvertently misclassified asymptomatic ONFH cases as non-ONFH controls.
ONFH clinical evaluation, functional assessment, and initial management
Clinical presentation varies, but most people with ONFH report hip pain (exacerbated by weight bearing), referred back and/or knee pain, decreased range of motion in the hip joint, gait abnormalities, and leg length discrepancies. ONFH can present in childhood with an inability to seat crosslegged or “ring seat.” The 2014 evidence-based report on SCD from the National Heart, Lung and Blood Institutes (NHLBI) recommends that all children and adults with intermittent hip pain undergo thorough clinical assessment and imaging (plain radiographs or MRI if needed) to rule out ONFH. Because ONFH often asymmetrically affects both hips and coexists with other joint involvement, anyone presenting with any of the aforementioned signs or symptoms should also undergo clinicoradiographic evaluation of the contralateral hip and bilateral shoulder joints to rule out multifocal OST. As demonstrated in our case, crutches can alleviate pain by enabling the patient to offload the symptomatic hip joint; however, such assistive devices can theoretically aggravate asymptomatic OST in other affected joints. ONFH diagnosis or suspicion thereof warrants a prompt referral to physical therapy/occupational therapy (PT/OT) for further evaluation and nonweight-bearing interventions.
Aguilar et al15 developed and validated the Children’s Hospital Oakland Hip Evaluation Scale (CHOHES)—a modified version of the Harris Hip Score—to assess symptoms and the functional status of people with sickle cell–related ONFH. Neumayr et al5 used the CHOHES tool in their prospective clinical trial, where they demonstrated that standardized physical therapy alone alleviated hip pain and debility at comparable rates with core decompression and physical therapy in a cohort of 46 adults with SCD and early-stage ONFH. In clinical practice, people’s experiences and results with PT/OT can vary widely, whereas some uninsured or underinsured patients lack access to physical/occupational health altogether. In such challenging cases, our PT/OT colleagues may recommend online stretching exercises or affordable community resources that our patients with early-stage ONFH can avail themselves of.
As our case demonstrates, ONFH diagnosis may be delayed for months and requires both clinical and radiographic criteria. Classification systems for ONFH diagnosis and staging include Ficat and Arlet, University of Pennsylvania (or Steinberg), Association Research Circulation Osseous, and Japanese Investigation Committee.16 Most studies cited in this review use the Steinberg classification (Table 1).17 Necrotic lesion volume and joint involvement primarily determine ONFH progression to femoral head collapse.18,19
University of Pennsylvania Classification of OST
| Stage | Criteria | |
|---|---|---|
| 0 | Normal or nondiagnostic radiograph, bone scan, and MRI | |
| I | Normal radiograph: abnormal bone scan and/or MRI | |
| A ‒ Mild | (<15% of head affected) | |
| B ‒ Moderate | (15%-30%) | |
| C ‒ Severe | (>30%) | |
| II | Lucent and sclerotic changes in femoral head | |
| A ‒ Mild | (<15%) | |
| B ‒ Moderate | (15%-30%) | |
| C ‒ Severe | (>30%) | |
| III | Subchondral collapse (crescent sign) without flattening | |
| A ‒ Mild | (<15% of articular surface) | |
| B ‒ Moderate | (15%-30%) | |
| C ‒ Severe | (>30%) | |
| IV | Flattening of femoral head | |
| A ‒ Mild | (<15% of surface and <2-mm depression) | |
| B ‒ Moderate | (15%-30% of surface or 2- to 4-mm depression) | |
| C ‒ Severe | (>30% of surface or >4-mm depression) | |
| V | Joint narrowing and/or acetabular changes | |
| A ‒ Mild |  | |
| B ‒ Moderate | ||
| C ‒ Severe | ||
| VI | Advanced degenerative changes | |
| Stage | Criteria | |
|---|---|---|
| 0 | Normal or nondiagnostic radiograph, bone scan, and MRI | |
| I | Normal radiograph: abnormal bone scan and/or MRI | |
| A ‒ Mild | (<15% of head affected) | |
| B ‒ Moderate | (15%-30%) | |
| C ‒ Severe | (>30%) | |
| II | Lucent and sclerotic changes in femoral head | |
| A ‒ Mild | (<15%) | |
| B ‒ Moderate | (15%-30%) | |
| C ‒ Severe | (>30%) | |
| III | Subchondral collapse (crescent sign) without flattening | |
| A ‒ Mild | (<15% of articular surface) | |
| B ‒ Moderate | (15%-30%) | |
| C ‒ Severe | (>30%) | |
| IV | Flattening of femoral head | |
| A ‒ Mild | (<15% of surface and <2-mm depression) | |
| B ‒ Moderate | (15%-30% of surface or 2- to 4-mm depression) | |
| C ‒ Severe | (>30% of surface or >4-mm depression) | |
| V | Joint narrowing and/or acetabular changes | |
| A ‒ Mild | | |
| B ‒ Moderate | ||
| C ‒ Severe | ||
| VI | Advanced degenerative changes | |
Reprinted from Lee et al17 with permission.
ONFH prognosis
Mont et al20 systematically reviewed 16 clinical studies that included 664 hips with asymptomatic ONFH and reported the risk factors for femoral head collapse. Key risk factors that predisposed to ONFH progression included the size and location of the necrotic lesion, ONFH stage at diagnosis, and associated clinical factors. When they stratified by underlying disease, SCD had the highest prevalence of progression to femoral head collapse (73%) compared with only 7% in systemic lupus erythematosus.
Hernigou et al21 consecutively recruited 121 ambulatory adults with SCD (42% female; mean age 26 years old; 40% Hb SS, 48% Hb SC, and 12% Hb S/β-thalassemia) with symptomatic ONFH in 1 hip and radiographically confirmed asymptomatic disease in the contralateral hip. All study participants underwent annual monitoring for clinical symptoms (onset of pain) and radiographic progression (defined as increase in Steinberg stage) over 10 to 20 years. At study entry, all 121 hips with asymptomatic ONFH were classified as Steinberg stages 0 to II; 93 hips (77%) progressed to advanced ONFH (Steinberg stage IV or higher) by the final mean follow-up at 14 years (Table 2).
ONFH progression to collapse
| Stage | Criteria for staging | No. of hips | |
|---|---|---|---|
| At initial visit | At final follow-up | ||
| 0 | Hip at risk with normal X-ray and MRI findings | 56 | 9 |
| I | Abnormal MRI findings, normal X-ray findings | 42 | 1 |
| II | Abnormal X-rays with sclerotic or cystic changes in the femoral head but no crescent line | 23 | 18 |
| III | Abnormal X-rays with a crescent sign and femoral head flattening ≤1 mm | 0 | 0 |
| IV | Collapse of the femoral head ≥1 mm without joint space narrowing | 0 | 2 |
| V | Joint space narrowing | 0 | 49 |
| VI | Advanced degenerative changes | 0 | 42 |
| Stage | Criteria for staging | No. of hips | |
|---|---|---|---|
| At initial visit | At final follow-up | ||
| 0 | Hip at risk with normal X-ray and MRI findings | 56 | 9 |
| I | Abnormal MRI findings, normal X-ray findings | 42 | 1 |
| II | Abnormal X-rays with sclerotic or cystic changes in the femoral head but no crescent line | 23 | 18 |
| III | Abnormal X-rays with a crescent sign and femoral head flattening ≤1 mm | 0 | 0 |
| IV | Collapse of the femoral head ≥1 mm without joint space narrowing | 0 | 2 |
| V | Joint space narrowing | 0 | 49 |
| VI | Advanced degenerative changes | 0 | 42 |
X-ray indicates plain radiographic imaging. Reprinted from Hernigou et al21 with permission.
Hernigou et al22 also studied 64 adults with SCD (48% female; mean age 29 years old; 64% Hb SS, 19% Hb SC, and 17% Hb S/β-thalassemia) without radiographic evidence of hip deformity at baseline for 13 to 20 years. Within 3 months of pain onset in 1 or both hips, study participants underwent routine plain radiographs or MRI to assess for progression to symptomatic ONFH. All 64 adults with SCD were recruited between June 1980 and December 1987, but the authors only incorporated MRI surveillance from 1985 to 1987. Thus, they may have missed very early-stage ONFH at study entry. Of the 92 hips that developed ONFH over the study period, 32 were Steinberg stage I (seen on MRI only), and 60 had plain radiographic evidence of ONFH per Steinberg criteria (43 stage II, 2 stage III, and 15 stage IV). Of the 77 hips without femoral head collapse (ie, Steinberg stages I-III), 65 (87%) progressed to Steinberg stage IV or higher within 5 years of diagnosis. On average, hips with stage I ONFH collapsed at 42 months, whereas those with stage II ONFH collapsed at 30 months. By the end of the follow-up period, 90 of 92 hips (98%) had collapsed, and 88 (96%) required surgery for intractable pain. The survivorship curves for all 92 hips, stratified by ONFH stage, is excerpted from the original paper (Figure 2).
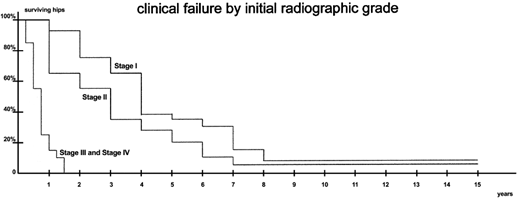
Time to femoral head collapse per ONFH stage at study entry. Adapted from Hernigou et al22 with permission.
Time to femoral head collapse per ONFH stage at study entry. Adapted from Hernigou et al22 with permission.
Itzep et al23 recently showed spontaneous ONFH regression in a retrospective cohort of 16 children with SCD (50% female; mean age 13.2 years old; 81% Hb SS, 13% Hb S/β0-thalassemia, and 6% Hb SC) who were followed at Texas Children’s Hospital Hematology Center from January 2006 to January 2018. Although all 16 children had similar Steinberg stages of ONFH at study entry, 5 children (33%) demonstrated ONFH regression from Steinberg stage IVB to IIB (group 1), whereas the remaining 11 showed stable to progressive ONFH from Steinberg stage IIIB to IVB (group 2). Compared with the children with stable or progressive ONFH, those with radiographic evidence of ONFH regression were younger (mean age 9.9 vs 14.6 years; P = .002). Of note, 5 of 16 children received physical therapy, but only 1 of the demonstrated ONFH regression. Five children also underwent core decompression with bone marrow aspirate injection without radiographic improvement.
Effects of HU on ONFH
HU and/or core decompression may have delayed ONFH progression in our patient’s left hip, but conflicting reports on the role of HU on the pathogenesis and prognosis of OST abound. Mahadeo et al24 implied that HU therapy predisposed to ONFH based on their study of 257 children, adolescents, and young adults with SCD (34% female; ages 10-21 years old; 69% Hb SS, 25% Hb SC, 5% Hb S/β0/+-thalassemia, and 1% Hb S/other) undergoing annual ONFH screening using plain radiographs from 2005 to 2010. They diagnosed ONFH in 32 people (12.5%; 50 of 64 hips); of these, 18 (56%) had bilateral hip disease. Similar to the CSSCD, they stratified ONFH prevalence by SCD genotype and found prevalence rates of 14.2% (26 of 182) in those with Hb SS/Hb Sβ0-thalassemia and 9.2% (6 of 65) in those with Hb SC. Only study participants with plain radiographic evidence of ONFH underwent pelvic MRI, presumably to distinguish between Steinberg stages 0 and 1; thus, the investigators may have misclassified others with early-stage ONFH as not having hip disease.
Mahadeo et al24 further analyzed data from study participants with Hb SS/Hb S/β0-thalassemia (n = 182), because they were 26 of 32 ONFH cases (81%), and ∼46% of them received HU therapy. Their univariate analyses showed that ONFH associated with older age, male sex, lower white blood cell count (WBC), higher hematocrit (Hct), lower lactate dehydrogenase (LDH), elevated creatinine, and HU exposure (which the investigators defined as receiving HU for >6 months). The multivariate regression model included HU, age, gender, ethnicity, SCD genotype, body mass index, creatinine, and hospitalizations for vaso-occlusive crises (VOCs); they excluded laboratory variables (Hct, LDH, MCV, and WBC) with collinearity with HU. The investigators used the expected laboratory values of lower WBC and absolute neutrophil counts, higher Hct and MCV, decreased reticulocyte counts, higher fetal Hb, and lower LDH in the HU-treated group as a proxy for HU adherence. They found independent associations between ONFH and HU (odds ratio [OR], 3.1; 95% confidence interval [95% CI]: 1.17, 8.41; P = .022), male gender (OR, 3.1; 95% CI: 1.05, 9.02; P = .04), and age (OR, 1.3; 95% CI: 1.07, 1.56; P = .008). The investigators clearly stated that their cross-sectional analysis precluded any conclusions on causality; other limitations included not factoring in duration of HU exposure and frequency of VOCs before HU initiation. Furthermore, they failed to report age at HU initiation and the number of patients on maximum tolerated dose (MTD) of HU (defined as the dose required to maintain absolute neutrophil count of 2000-4000/µL per NHLBI criteria).25
Other studies found no association between HU exposure and ONFH.26-28 Gupta and Adekile29 conducted a longitudinal prospective study of ONFH incidence and progression in a cohort of 30 pediatric subjects with sickle cell anemia (mean age 9.8 ± 3.5 years old; 23 Hb SS and 7 Hb S/β0-thalassemia) from Kuwait. Of these, 17 underwent serial hip MRI assessments over 1 to 4 years. They found baseline ONFH in 11 of 34 hips (32.4%) examined; by the end of the study period, 22 (64.7%) had developed ONFH. Given the mild clinical phenotype and high baseline fetal Hb levels of this patient population—attributed to the Arab/India haplotype—only 2 children were started on HU for frequent VOCs at ∼6 months before their first hip MRI. The study participants with progressive ONFH had frequent VOCs, whereas 2 of 3 with stable ONFH had started HU before their first hip MRI. This latter observation prompted additional investigation into the role of HU in delaying the onset or progression of ONFH in children with SCD.
In a follow-up study, Adekile et al30 recently reported on the prevalence, incidence, and progression of ONFH in a prospective cohort of 40 Kuwaiti children and adolescents with SCD (mean age 12.9 ± 4.2 years old at study entry; 45% Hb SS, 47.5% Hb S/β0-thalassemia, and 7.5% Hb SD) who received HU over 1 to 15 years. Hip MRI studies obtained before HU initiation confirmed ONFH at varying stages in 11 people (27.5%). Study participants with ONFH before HU initiation were statistically significantly older than those without ONFH (15.3 ± 3.7 vs 11.9 ± 4.0 years old; P = .02). Hip MRIs, obtained annually, showed incident ONFH in 2 of 29 (6.9%) without ONFH at study entry. Five of 11 with baseline ONFH (45.5%) remained stable, another 5 (45.5%) progressed, and 1 (9%) showed radiographic improvement on hip MRI. Adekile et al30 and Gupta and Adekile29 surmised that HU may protect against ONFH onset and retard its progression in children and adolescents with SCD.
Analysis of variance test showed that reticulocyte count statistically significantly differed between those without ONFH (3.8% ± 2.4%) compared with those with stable (6.8% ± 1.4%) or progressive (6.3% ± 3.5%) disease. Concentrations of Hb/Hct, fetal Hb, and LDH were similar across all groups. Multiple ordinal regression analyses showed significant associations between ONFH and pre-HU age, platelets, and reticulocyte counts. Contrary to the CSSCD, baseline ONFH prevalence and severity did not differ based on SCD genotype or coexistent thalassemia trait. Unlike our OSHPD study8 and other reports,31 ONFH did not significantly associate with frequent VOCs or history of ACS (relative risk [RR], 1.2; P > .05). Of note, this Kuwaiti SCD cohort mostly had the Arab/India haplotype (high baseline Hb and fetal Hb levels) that manifests as a more vaso-occlusive/viscosity SCD subphenotype.31 The association between ONFH and reticulocytosis suggests that hemolysis may play a role in ONFH development in Kuwaiti children and adolescents with SCD.
In the case series by Itzep et al,23 all 16 children were on comparable HU doses (at MTD) for similar treatment durations over the observation period. The 5 children with ONFH regression (group 1) were younger and had lower mean MCV values compared with the 11 children with stable to progressive ONFH (group 2); however, mean Hb, fetal Hb, whole-blood viscosity, red cell deformability, and percentage dense RBCs were similar in both groups. Although a small case series, this study offers a granular insight into HU effect on ONFH. Although poorer HU adherence could have caused the lower MCV in group 1, all other measures of HU effect were comparable between groups 1 and 2. The authors failed to report the prevalence of coinherited α-thalassemia in their study cohort, but group 1 had lower mean MCV and lower mean Hb level, which argues against concomitant α-thalassemia. In summary, ONFH prognosis in this small cohort of children with SCD seems independent of HU exposure.
Lemonne et al26 previously compared hematologic and hemorheologic parameters in a cohort of 97 adults with sickle cell anemia stratified into those with OST (OST+; n = 30; 63% females; mean age 39.3 ± 13.1 years old) and without OST (OST−, n = 67; 52% females). In addition to the typical OST risk factors of older age, coinherited thalassemia, more frequent VOCs within the past year, and higher baseline Hb, the OST+ subgroup also had higher RBC deformability and aggregation. After excluding a subset of 18 sickle cell anemia adults on HU, the investigators still found that OST significantly associated with age (OR, 1.06; 95% CI: 1.101, 1.12; P < .05), Hb (OR, 2.24; 95% CI: 1.19, 4.18; P < .05), and RBC deformability (OR, 1.15; 95% CI: 1.01, 1.33; P < .05). They explained that RBC deformability resulted in more frequent vaso-occlusion, because irregularly shaped and deformable sickle RBCS adhered more to the vascular endothelium than to rigid, irreversibly sickled RBCs. They attributed the association between increased RBC deformability and OST to possible confounding from coinherited α-thalassemia, because deformed RBCs were more prevalent in those with α-thalassemia (0.18 ± 0.05) compared with those without (0.15 ± 0.06; P < .05). Despite higher baseline Hb levels in the OST+ group, the investigators reported no significant increases in blood viscosity.
Lemonne et al32 subsequently measured HU effect on blood viscosity and RBC rheology (ie, RBC deformability and aggregation as previously described) in a cohort of 24 adults with sickle cell anemia (mean age 38.3 ± 11.8 years old; 42% female; 50% with 1 α-gene deletion). They demonstrated that, despite a gradual increase in Hb/Hct levels with HU, blood viscosity remained unchanged, and RBC rheologic parameters improved. In a follow-up study of a similar cohort of 28 adults (mean age 37.0 ± 11.86 years old; 46% females; 50% ∝3.7 deletion) who were initiated on HU at baseline and maintained on a mean dose of HU 15.7 ± 5.4 mg/kg per day, Lemonne et al33 confirmed sustained increases in Hb, fetal Hb, and mean corpuscular volume; stable blood viscosity and RBC aggregation; and increased RBC deformability over the 2-year study duration.
The OSHPD study gave us the unique advantage of studying the effect of HU on ONFH incidence. HU received Food and Drug Administration approval for treatment of adults with severe SCD in 1998; the 22-year study duration of our observational study spanned the decades before and after this period. We found an overall ONFH incident rate of 2.37 per 100 person-years (95% CI: 2.18, 2.56) in the pre-HU era (1991-2000) compared with 1.52 per 100 person-years (95% CI: 1.41, 1.63) in the post-HU era (2001-2013). The incident rate per age decile was excerpted from our original manuscript (Table 3). Because the OSHPD lacks medication prescription data, we could not verify that people with SCD were routinely prescribed HU after 1998, and we could not prove adherence or MTD escalation. Rather than directly associate the reduced ONFH incidence to concomitant HU therapy, we speculated that HU likely reduced emergency room visits and hospitalizations for VOCs or ACS, which in turn, decreased our ability to detect ONFH discharge diagnoses.
ONFH incident rates pre- and post-HU era
| Age category (y) | Pre-HU (1991-2000) | Post-HU (2001-2013) | ||||
|---|---|---|---|---|---|---|
| ONFH cases | Incident rate* | 95% CI | ONFH cases | Incident rate* | 95% CI | |
| <10 | 46 | 0.85 | 0.63, 1.10 | 31 | 0.46 | 0.32, 0.64 |
| 10 to <20 | 137 | 2.65 | 2.23, 3.12 | 145 | 1.27 | 1.07, 1.49 |
| 20 to <30 | 151 | 2.52 | 1.93, 2.70 | 236 | 2.11 | 1.86, 2.40 |
| 30 to <40 | 133 | 2.48 | 2.08, 2.93 | 158 | 1.66 | 1.42, 1.93 |
| 40 to <50 | 73 | 2.58 | 2.04, 3.23 | 110 | 1.44 | 1.19, 1.73 |
| 50 to <60 | 38 | 4.43 | 3.18, 6.02 | 69 | 1.78 | 1.39, 2.23 |
| ≥60 | 10 | 6.51 | 3.31, 11.6 | 19 | 1.94 | 1.21, 2.98 |
| Total | 588 | 2.37 | 2.18, 2.56 | 768 | 1.52 | 1.41, 1.63 |
| Age category (y) | Pre-HU (1991-2000) | Post-HU (2001-2013) | ||||
|---|---|---|---|---|---|---|
| ONFH cases | Incident rate* | 95% CI | ONFH cases | Incident rate* | 95% CI | |
| <10 | 46 | 0.85 | 0.63, 1.10 | 31 | 0.46 | 0.32, 0.64 |
| 10 to <20 | 137 | 2.65 | 2.23, 3.12 | 145 | 1.27 | 1.07, 1.49 |
| 20 to <30 | 151 | 2.52 | 1.93, 2.70 | 236 | 2.11 | 1.86, 2.40 |
| 30 to <40 | 133 | 2.48 | 2.08, 2.93 | 158 | 1.66 | 1.42, 1.93 |
| 40 to <50 | 73 | 2.58 | 2.04, 3.23 | 110 | 1.44 | 1.19, 1.73 |
| 50 to <60 | 38 | 4.43 | 3.18, 6.02 | 69 | 1.78 | 1.39, 2.23 |
| ≥60 | 10 | 6.51 | 3.31, 11.6 | 19 | 1.94 | 1.21, 2.98 |
| Total | 588 | 2.37 | 2.18, 2.56 | 768 | 1.52 | 1.41, 1.63 |
Reprinted from Adesina et al8 with permission.
Incident rate per 100 person-years.
Surgical outcomes in ONFH
Core decompression—composed of drilling or multiple fenestrations through the femoral neck to stimulate revascularization of the necrotic femoral head—may alleviate symptoms in early-stage ONFH with small necrotic volumes.34 Many people with SCD complicated by symptomatic ONFH are diagnosed at later stages when nonweight bearing, physical therapy, or core decompression offers little to no relief. Early orthopedic surgery evaluations are appropriate for people with intractable pain and/or impaired mobility related to their underlying ONFH. Historically, poor perioperative outcomes and suboptimal postoperative pain relief dissuaded many people with SCD and advanced ONFH from pursuing hip replacement surgery.35,36
Vichinsky et al37 reported a 67% perioperative complication rate in 118 adults with sickle cell anemia undergoing 138 orthopedic procedures (127 in Hb SS patients and 11 in Hb S/β0-thalassemia patients; 55% female; mean age 26 years old). Study participants either received aggressive perioperative transfusions to achieve goal Hb S < 30% and Hb 9 to 11 g/dL (groups 1 or 3) or conservative simple transfusions to increase Hb levels to 9 to 11 g/dL (groups 2 or 4). Other than Hb S levels and history of cardiac disease, demographics, clinical history, and baseline laboratory values were similar across the randomized groups (ie, groups 1 and 2). Serious complication rates for all 138 surgeries, including hip coring (low risk) and hip arthroplasty (intermediate risk), were excerpted from the paper and summarized in Table 4.
Perioperative complications of orthopedic procedures (overall) compared with hip replacement surgery and hip coring
| Any perioperative complications* | Total group | Hip replacement | Hip coring | |||
|---|---|---|---|---|---|---|
| n = 138 | % | n = 52 | % | n = 22 | % | |
| Intraoperative/PACU | 73 | 53 | 48 | 92 | 5 | 23 |
| Excessive blood loss (>10% excessive blood loss) | 61 | 44 | 37 | 71 | 2 | 9 |
| Other | 27 | 20 | 11 | 21 | 4 | 18 |
| ACS | 16 | 12 | 8 | 15 | 3 | 14 |
| Miscellaneous postoperative | 18 | 13 | 12 | 23 | 1 | 5 |
| Fever/infection | 19 | 14 | 8 | 15 | 1 | 5 |
| VOE | 13 | 9 | 6 | 12 | 2 | 9 |
| Neurologic | 5 | 4 | 2 | 4 | 1 | 5 |
| Renal | 1 | 1 | 1 | 2 | 0 | 0 |
| Death | 2 | 1 | 1 | 2 | 0 | 0 |
| Sickle cell event (ACS/VOE) | 23 | 17 | 10 | 19 | 4 | 18 |
| Any serious complication | 92 | 67 | 50 | 96 | 10 | 45 |
| Any perioperative complications* | Total group | Hip replacement | Hip coring | |||
|---|---|---|---|---|---|---|
| n = 138 | % | n = 52 | % | n = 22 | % | |
| Intraoperative/PACU | 73 | 53 | 48 | 92 | 5 | 23 |
| Excessive blood loss (>10% excessive blood loss) | 61 | 44 | 37 | 71 | 2 | 9 |
| Other | 27 | 20 | 11 | 21 | 4 | 18 |
| ACS | 16 | 12 | 8 | 15 | 3 | 14 |
| Miscellaneous postoperative | 18 | 13 | 12 | 23 | 1 | 5 |
| Fever/infection | 19 | 14 | 8 | 15 | 1 | 5 |
| VOE | 13 | 9 | 6 | 12 | 2 | 9 |
| Neurologic | 5 | 4 | 2 | 4 | 1 | 5 |
| Renal | 1 | 1 | 1 | 2 | 0 | 0 |
| Death | 2 | 1 | 1 | 2 | 0 | 0 |
| Sickle cell event (ACS/VOE) | 23 | 17 | 10 | 19 | 4 | 18 |
| Any serious complication | 92 | 67 | 50 | 96 | 10 | 45 |
Complications associated with transfusions were excluded. Percentages are the proportion of operations in each group with a complication. PACU, postanesthesia care unit. Adapted from Vichinsky et al37 with permission.
Includes preoperative period after transfusion.
Issa et al38 retrospectively studied surgical outcomes in 32 people with sickle hemoglobinopathies (10 sickle cell trait and 22 SCD [genotype not specified]; mean age 37 years old; mean follow-up duration 7.5 years) undergoing THA with cementless prostheses for 42 hips. All SCD patients received aggressive perioperative RBC transfusion with goal Hb of 9 to 11 g/dL and Hb S < 30% as described in the study by Vichinsky et al.37 Compared with 87 non-SCD patients (102 hips at similar ONFH stage, necrotic lesion size, and femoral head collapse), SCD patients had similar cementless prostheses survival, postoperative pain, range of motion, function, quality of life, and radiographic findings.
Ilyas et al39 also reported favorable surgical outcomes in 101 people with SCD (all Hb SS; 51% female; mean age 25 years old; follow-up duration 5-17 years) who accounted for 133 hips with ONFH (Steinberg stage IV or higher) and underwent noncemented THA. The investigators carefully monitored hydration, oxygenation, and analgesia for all participants in the perioperative setting. They preemptively administered gentamicin and cefazolin and then continued cefazolin postoperatively until intraoperative bone cultures were resulted. If needed, study participants received RBC transfusions to reach/maintain an Hb goal of 10 g/dL in the perioperative period. Table 5 summarizes updated rates of perioperative complications of THA in sickle cell–related ONFH adapted from the original paper. The investigators reported almost 97% implant survivorship at 10 years and 94% at 15 years; surgical revisions for primary THA failure (aseptic loosening, dislocation, or infection) were associated with an almost 98% survival at mean follow-up of 16.5 years.
Rates of perioperative complications in 101 adults (133 hips) undergoing THA for sickle cell–related ONFH
| Type of complication | Total hips (n = 133) | Rate (%) |
|---|---|---|
| Intraoperative proximal femoral fracture | 6 | 4.5 |
| Vaso-occlusive sickle cell crisis | 12 | 9.0 |
| Superficial wound infection | 4 | 3.0 |
| Deep wound infection | 5 | 3.8 |
| Heterotopic ossification (grade IV) | 5 | 3.8 |
| Hip instability | 1 | 0.8 |
| Acetabular protrusion | 2 | 1.5 |
| Aseptic failure of cup | 1 | 0.8 |
| Sciatic nerve plasy | 2 | 1.5 |
| Type of complication | Total hips (n = 133) | Rate (%) |
|---|---|---|
| Intraoperative proximal femoral fracture | 6 | 4.5 |
| Vaso-occlusive sickle cell crisis | 12 | 9.0 |
| Superficial wound infection | 4 | 3.0 |
| Deep wound infection | 5 | 3.8 |
| Heterotopic ossification (grade IV) | 5 | 3.8 |
| Hip instability | 1 | 0.8 |
| Acetabular protrusion | 2 | 1.5 |
| Aseptic failure of cup | 1 | 0.8 |
| Sciatic nerve plasy | 2 | 1.5 |
Adapted from Ilyas et al39 with permission.
Lastly, Farook et al40 reported outcomes in 30 adults with SCD (mean age 36.7 years old; 60 females; 80% Hb SS, 13% Hb SC, and 7% Hb S/β-thalassemia or other Hb variant) with Steinberg stage IV or higher ONFH affecting 34 hips. Approximately 76% of the THAs incorporated uncemented hip implants, whereas the rest used hybrid or cemented prostheses. At a mean follow-up duration of 10.5 years, the authors reported prosthetic joint infections (5.8%), aseptic acetabular failure (11.7%), and surgical revisions (17.6%) without any cases of aseptic femoral failure.
Conclusion
ONFH affects ∼20% to 50% of people with SCD and causes significant morbidity as it inevitably progresses to femoral head collapse. Frequent VOEs and ACS remain the most consistent comorbidities of ONFH. To date, no studies have proven a causal effect between HU and ONFH. Evaluation of nonsurgical interventions (radiofrequency ablation of femoral joint nerves, massage therapy, acupuncture, exercise, or arthroscopic joint injections) or surgical adjuncts (bone graft, autologous stem cell infusions, etc) is beyond the scope of this review. An evidence-based approach to perioperative SCD management with appropriate hydration, oxygenation, RBC transfusions, and pain control—along with advanced surgical techniques and use of noncemented hip prostheses—increases the likelihood of favorable postoperative outcomes and improved quality of life41 for people undergoing hip arthroplasty for sickle cell–related ONFH.
Correspondence
Oyebimpe O. Adesina, University of Washington, Seattle Cancer Care Alliance, 825 Eastlake Ave E, MS CE3-300, Seattle, WA 98109; e-mail: adesina@uw.edu.
