
Abstract
Before the advent of effective iron chelation, death from iron-induced cardiomyopathy occurred in the second decade in patients with transfusion-dependent chronic anemias. The advances in our understanding of iron metabolism; the ability to monitor iron loading in the liver, heart, pancreas and pituitary; and the availability of several effective iron chelators have dramatically improved survival and reduced morbidity from transfusion-related iron overload. Nevertheless, significantly increased survival brings about new complications such as malignant transformation resulting from prolonged exposure to iron, which need to be considered when developing long-term therapeutic strategies. This review discusses the current biology of iron homeostasis and its close relation to marrow activity in patients with transfusion-dependent anemias, and how biology informs clinical approach to treatment.
Learning Objectives
Discuss the mechanism of nonregulated entry of iron into organs and the importance of this in clinical management
Discuss the critical role of ineffective erythropoiesis and marrow activity organ distribution of iron and iron toxicity
Discuss the importance of continual levels in preventing and treating cardiac and endocrine organ iron overload
Discuss the different consequences of iron toxicity now that survival is better and patients are exposed for decades
Introduction
Blood transfusion continues to play a very important role in modern medicine, at least in countries with more advanced medical technology, where there is good blood supply and the complications of transfusion such as infection and transfusion reaction have been reduced to very low rates. However, iron overload secondary to transfusions remains a significant problem. Considering that each milliliter of packed red cells contains 0.8 mg iron and that humans have no means to lose iron other than the approximately 2 mg/day lost from desquamated oral and intestinal epithelia,1 patients who receive multiple transfusions can rapidly become iron loaded. From experience with thalassemia, in which the causal relationship of iron overload to mortality has been clearly demonstrated,2,3 we know that iron overload is toxic and can be lethal. In the 1950s, before patients were regularly transfused, the median survival from thalassemia major was around 4 to 5 years of age. Once transfusions were used to maintain hemoglobin levels at least in the 7 to 8 g/dL range, survival improved in these children, but they still succumbed to heart and endocrine failure at around 15 years of age. The mental picture of a child who has gray-blue skin, has not gone through puberty, has “bronze” diabetes and heart failure is striking, and has influenced our thinking about iron overload of all types. In fact, these toxic effects of iron overload on the heart can now be largely prevented or reversed, and a less predictable but still reasonable chance of protecting the endocrine organs and improving function is now a possibility. Patients with thalassemia major are currently living into their 50s and 60s, but cardiac and endocrine failure are being replaced by malignant transformation, as a consequence of decades of exposure to low or moderate levels of oxidatively active iron (reviewed in Coates et al4 ). Thanks to our ability to monitor iron loading in specific organs and to significant advances in our understanding of iron homeostasis, we now know that the toxicity from iron overload is not the same for all anemia disorders, and in fact highly depends on bone marrow activity, specific organ distribution of iron overload, and duration of exposure. Although it is clear that the rate of total body iron loading is linearly related to the number of transfusions,5 the iron loading rates of the pancreas, pituitary gland, and heart are quite different and highly dependent on organ-specific iron transport differences and bone marrow activity. Similarly, the rate of iron unloading in the liver is much faster than the rate of unloading from the endocrine organs and heart. This results in patients having nearly normal total body iron stores (liver iron concentration; LIC) after chelation, but substantial residual iron loading in the endocrine organs and heart.
We discuss the current understanding of iron homeostasis in humans and how this new knowledge has informed our approach to clinical management of transfusional iron overload.
Case: a 28-year-old patient with thalassemia major with transfusional iron overload
MC is a 28-year-old male Asian college student with transfusion-dependent β-thalassemia major who was started on transfusions at around 6 months of age. His hemoglobin levels have generally been kept in a 7 to 8 g/dL range. He was started on iron chelation with deferoxamine at 5 years of age. His family moved to the United States when he was 18 years of age. He has received transfusions approximately every 3 weeks, and has been receiving chelation with various combinations of deferasirox (DFX), deferoxamine, and deferiprone (DFP). For the most recent 3 to 5 years, he has been adherent to his chelation therapy. He has severe osteoporosis (lumbar T score, −6.4), hypothyroidism, very low testosterone, type 2 diabetes, and a physical examination consistent with growth hormone deficiency. A magnetic resonance imaging scan (MRI) of his liver performed the year before being followed at our center showed a LIC of 2.5 mg/g dry weight liver (near normal). Studies performed at our center are shown in Table 1. His liver iron of 2.9 mg/g liver dry weight in 2017 suggested that his total body iron burden was fairly well controlled and that he is sensitive to chelation. However, his pituitary iron was 8 standard deviations above the mean, his pituitary volume was 3.8 standard deviations below the mean, and his pancreatic iron was 5 times the normal, well into the range associated with significant glucose intolerance. His cardiac T2* was 15 ms, indicating moderate iron loading. He was taking DFX at about 70% the maximum dose once a day when we met him in 2017. The main issue was to determine how to reduce the substantial iron loading in his heart and endocrine organs without putting him at risk for chelator toxicity associated with the relatively low total body iron.
Case: patient with thalassemia major with iron overload
| Iron loaded patient with thalassemia major | ||||
|---|---|---|---|---|
| Target | 2017 | 2018 | 2019 | |
| Hemoglobin, gm/dL | >10 g/dL | 11.6 | 12.0 | 11.5 |
| Ferritin, ng/dL | <200 ng/dL | 3000 | 2500 | 1750 |
| t-SAT, % | <50% | 60% | 45% | 70% |
| DFX, mg† | 22 mg/kg/day | 1440 daily | 720 twice daily | 720 twice daily |
| Liver Fe, mg/g‡ | <1.5 mg/g | 2.9 | 3.0 | 2.9 |
| Pancreas R2* (Hz) | <27 Hz | 240 | 160 | 90 |
| Pit Fe Z score | 0 | 8.2 | 7 | 6.5 |
| Pit vol Z score | 0 | −3.8 | −3.6 | −3.6 |
| Cardiac T2*, ms | >30 ms | 15 | 17 | 24 |
| Cardiac iron, mg/g | <0.71 mg/g | 1.65 | 1.42 | 0.93 |
| LVEF, % | >56% | 58 | 60 | 58 |
| Iron loaded patient with thalassemia major | ||||
|---|---|---|---|---|
| Target | 2017 | 2018 | 2019 | |
| Hemoglobin, gm/dL | >10 g/dL | 11.6 | 12.0 | 11.5 |
| Ferritin, ng/dL | <200 ng/dL | 3000 | 2500 | 1750 |
| t-SAT, % | <50% | 60% | 45% | 70% |
| DFX, mg† | 22 mg/kg/day | 1440 daily | 720 twice daily | 720 twice daily |
| Liver Fe, mg/g‡ | <1.5 mg/g | 2.9 | 3.0 | 2.9 |
| Pancreas R2* (Hz) | <27 Hz | 240 | 160 | 90 |
| Pit Fe Z score | 0 | 8.2 | 7 | 6.5 |
| Pit vol Z score | 0 | −3.8 | −3.6 | −3.6 |
| Cardiac T2*, ms | >30 ms | 15 | 17 | 24 |
| Cardiac iron, mg/g | <0.71 mg/g | 1.65 | 1.42 | 0.93 |
| LVEF, % | >56% | 58 | 60 | 58 |
LVEF, left ventricular ejection fraction by MRI; Pit, pituitary; t-SAT, transferrin saturation.
DFX as tablets (JadeNu). Total dose change to twice daily in 2017 (off-label use)
Based on R2*.
Iron balance and toxicity in humans
Iron homeostasis
To understand how to best treat this patient without putting him at risk from overchelation, we needed to delve into the details of iron homeostasis. Advances in our understanding of iron transport coupled with serial MRI measures of tissue iron during loading and unloading demonstrate that iron transport occurs at significantly different rates in different tissues.6,7 Liver iron concentration is highly correlated (r2 = 0.98; P < .001) with total body iron5 ; however, the correlation among LIC and pancreatic, pituitary, or cardiac iron concentrations is very poor, indicating that factors other than total body iron control iron trafficking in these organs. In response to intensive iron chelation, the LIC can be reduced by 50% in 4 to 6 months, whereas it takes about 14 months to remove half of the iron from the heart.6 Pancreatic loading precedes cardiac loading. These significant differences in loading and unloading between various tissues (Figure 1), derived from serial MRI measurements of iron-loading in humans, are consistent with very elegant biochemistry studies of iron regulation during the last 2 decades (reviewed in Coates8 ) and have a direct effect on the diagnosis, monitoring, and treatment of transfusion-dependent patients with iron overload.
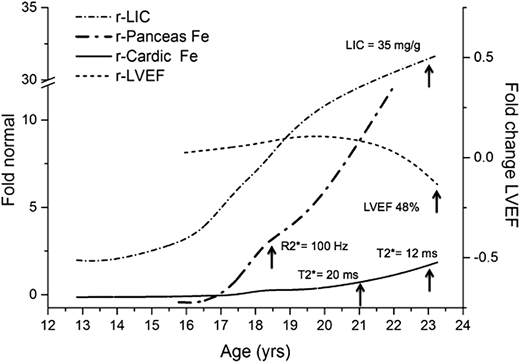
Temporal partitioning of total body iron (LIC) into pancreas and heart in a chronically transfused patient who was minimally adherent to chelation. Measurements of the organ iron by MRI iron are plotted (left axis) as fold change relative to normal. Note that the LIC is already 4 times normal at age 13 years. The pancreas iron (nl = 27 Hz) reaches the level associated with glucose intolerance (100 Hz) at 18.8 years of age, well before the cardiac T2* drops below 20 ms at about 21 years. The cardiac iron starts to rise at 18 years. The LVEF by MRI drops at age 23 years (from Coates and Wood,24 with permission).
Temporal partitioning of total body iron (LIC) into pancreas and heart in a chronically transfused patient who was minimally adherent to chelation. Measurements of the organ iron by MRI iron are plotted (left axis) as fold change relative to normal. Note that the LIC is already 4 times normal at age 13 years. The pancreas iron (nl = 27 Hz) reaches the level associated with glucose intolerance (100 Hz) at 18.8 years of age, well before the cardiac T2* drops below 20 ms at about 21 years. The cardiac iron starts to rise at 18 years. The LVEF by MRI drops at age 23 years (from Coates and Wood,24 with permission).
Case in point.
These differences in iron removal rate explain why the total body iron levels were rapidly brought down to near normal in the case here, as indicated by the LIC of 2.9 mg/g, while still having very high levels of iron in the pituitary and pancreas and moderate iron levels in the heart. The LIC response indicates that the patient is sensitive to DFX at this less-than-maximal dose.
Iron balance is maintained entirely through the modulation of dietary iron absorption in the duodenum and the recycling of about 25 mg of iron per day from phagocytosis of senescent autologous or transfused red blood cells by macrophages. The reclaimed iron in the macrophage is in the ferrous state (Fe+2) and is referred to as labile cellular iron (LCI). Iron as Fe+2 is highly reactive and causes protein and DNA damage. It is also the only form of iron that can pass through iron transporters such as ferroportin. LCI triggers the production of ferritin in the cell, is taken up and converted to nonreactive Fe+3 by ferritin, or is exported to the plasma via ferroportin, the only known cellular iron exporter. Ferritin is intracellular but can leak into the plasma when cell membranes are damaged. Thus, serum/plasma ferritin levels are a rough measure of iron loading, but are also sensitive to inflammation and cell membrane leak. Hemosiderin, which is made up of aggregates of ferritin, is the primary species that is detected by MRI (reviewed in Coates8 ). Importantly, MRI does not report ferritin itself or Fe+2, although this species is likely high in proximity to very high levels of the aggregated forms of Fe+3 detected by MRI.
Fe+3 circulates in plasma bound to transferrin, and normally, about 30% of the iron binding sites of the circulating transferrin are occupied. The complex enters cells by receptor-mediated endocytosis via the transferrin receptor-1, TfR1, present on all cells. TfR1 transcription decreases when intracellular Fe+2/LCI increases, thereby preventing cellular iron overload, at least by TfR1-mediated mechanisms. However, when total body iron is significantly elevated, as it is the case in multiply transfused patients, the transferrin iron-binding ability is exceeded (transferrin saturation > 60%-99%), and circulating nontransferrin-bound iron (NTBI) appears in the plasma (Figure 2A). NTBI rises markedly when the transferrin saturation reaches about 70%, and a highly reactive Fe+2 subspecies of NTBI called labile plasma iron (LPI) increases concomitantly. LPI can enter cells through ion transporters that are normally designed to carry divalent cations such as zinc and calcium. These ion transporters are generally not regulated by intracellular iron concentration, bypassing the TfR1 regulatory mechanisms; thus, iron loading proceeds, even when cytosolic iron levels are very high. The iron transport through these ion transporters is organ-dependent, which may explain the different organ rates of loading observed by serial MRI (liver>pancreas>heart).8 Transferrin receptor-2 (TfR2) is present only in liver, is not sensitive to cytosolic iron, and functions as the plasma iron sensor. It allows loading of iron into the liver, even if hepatic iron is high, and may account in part for why the liver loads so quickly.
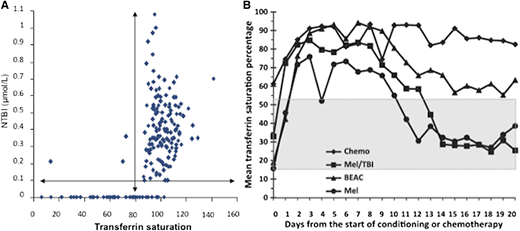
(A) Relation of NTBI to transferrin saturation. (B) Dramatic increase in transferrin saturation as a surrogate for NTBI/LPI when erythropoiesis is stopped by pretransplant chemotherapy, with normalization coincident with engraftment. “MEL” is a mild regimen that recovers quickly, “Chemo” is an intense regimen that recovers late (from Sahlstedt et al12 and Sahlstedt et al,28 with permission).
(A) Relation of NTBI to transferrin saturation. (B) Dramatic increase in transferrin saturation as a surrogate for NTBI/LPI when erythropoiesis is stopped by pretransplant chemotherapy, with normalization coincident with engraftment. “MEL” is a mild regimen that recovers quickly, “Chemo” is an intense regimen that recovers late (from Sahlstedt et al12 and Sahlstedt et al,28 with permission).
Importantly, this means that the heart, pancreas, and pituitary load with iron only under pathologic conditions when the transferrin binding capacity has been exceeded and there is circulating NTBI/LPI. Importantly, NTBI/LPI levels can go to zero as soon as there is circulating chelator, blocking further loading through ion transporters. This means that a chelator should be circulating at all times. It takes months or longer for iron to appear in the pancreas by MRI (Figure 1); therefore, presence of excess iron in the pancreas, pituitary, or cardiac iron means that the patient has been exposed to prolonged elevations of reactive NTBI/LPI. Accumulation in these organs can also mean the patient is only taking their chelator intermittently.
Case in point.
The elevated cardiac, pituitary and pancreatic iron levels indicate that our patient was exposed to high NTI/LPI levels for a long time. As the LIC is near normal, it indicates that the patient has been taking at least some of the prescribed chelator, but the high pancreas iron suggests that he may only be taking the chelator intermittently or at least has significant times during the day when chelator levels are low and NTBI/LPI are elevated. The reduction in total iron (LIC) is a good sign, but one needs to keep in mind that iron removal is much slower in the heart and endocrine organs.
Export of iron from enterocytes and macrophages into the plasma is regulated by hepcidin, a 25-amino acid, defensin-like peptide that is made in the liver and binds to ferroportin, causing its internalization and degradation. Thus, high hepcidin levels block movement of iron absorbed by enterocytes and stored by macrophages to plasma. Iron overload and inflammatory states elevate hepcidin.9,10 Conversely, hypoxia, anemia, and erythropoiesis reduce hepcidin production, which increases iron absorption and transport of Fe+2 into the plasma.11 Thus, the state of bone marrow activity has a significant effect on hepcidin production, and consequently iron loading.
Marrow activity has also a dramatic effect on the levels of reactive NTBI/LPI. As the marrow uses about 25 mg of iron a day, the levels of NTBI are much higher in the presence of marrow failure or ineffective erythropoiesis (Figure 3). This relationship can also be clearly seen when marrow activity is reduced by ablative chemotherapy in preparation for marrow transplant. NTBI reported by transferrin saturation goes up dramatically during the week after conditioning chemotherapy and drops back to normal when erythropoietic activity returns after engraftment12 (Figure 2B).

Iron loading in the patients with ineffective or no erythropoiesis (black) or effective erythropoiesis (red). All groups load the liver equally. The heart and pancreas, which primarily load when NTBI/LPI is high, have significantly less loading for disorders with effective erythropoiesis. The dashed line is the normal level. CDA, congenital dyserythropoietic anemia; DBA, Diamond Blackfan anemia; PK, pyruvate kinase deficiency; SCD, sickle cell anemia; TM, thalassemia major (from Coates8 and Berdoukas et al,13 with permission).
Iron loading in the patients with ineffective or no erythropoiesis (black) or effective erythropoiesis (red). All groups load the liver equally. The heart and pancreas, which primarily load when NTBI/LPI is high, have significantly less loading for disorders with effective erythropoiesis. The dashed line is the normal level. CDA, congenital dyserythropoietic anemia; DBA, Diamond Blackfan anemia; PK, pyruvate kinase deficiency; SCD, sickle cell anemia; TM, thalassemia major (from Coates8 and Berdoukas et al,13 with permission).
This dependence of NTBI/LPI on marrow activity is of great importance. In diseases with ineffective erythropoiesis (thalassemia, congenital dyserythropoietic anemia, low-grade myelodysplastic syndromes) or no erythropoiesis (Diamond Blackfan Anemia, aplastic anemia), patients have very high levels of NTBI/LPI because the iron is not being used to make more red cells. Thus, significant loading of the heart and endocrine organs13 and greater organ toxicity are observed in these patients than in those with chronic anemia and effective erythropoiesis (sickle cell disease, pyruvate kinase deficiency). Furthermore, patients with ineffective erythropoiesis hyperabsorb iron from their diet and become iron loaded even in the absence of transfusion.
Iron toxicity
The toxicity from iron is a result of the Fe+2 reactive forms of iron that can react with oxidants to rapidly damage proteins and DNA, permanently altering protein structure and genetic material. In conditions of iron overload when NTBI and LPI/LCI levels are high, and during inflammation when high levels of reactive oxygen species are produced, severe oxidant damage to tissues can occur as a result of the interactions of oxidants with Fe+2 (reviewed in Koskenkorva-Frank et al14 ).
The notion that NTBI/LPI is the toxic form of iron is consistent with clinical evidence. NTBI/LPI represents the pool of iron that is immediately accessible to chelators, and NTBI/LPI levels in the blood can be reduced to near zero within minutes to hours of starting chelation with deferoxamine.15 Plasma NTBI levels drop after any chelator is administered and are the mirror image of the plasma chelator levels for DFX and DFP as well. When patients with severe myocardial iron loading and decreased left ventricular function were treated with continuous iron chelation therapy, normalization of ejection fraction occurred within 3 months of starting chelation, even though the cardiac iron levels detected by MRI remain remarkably elevated. Thus, chelating NTBI/LPI was sufficient to improve cardiac function,6 and circulating chelator can be protective, even before tissue levels of iron have been significantly reduced. We have personal experience with a 32-year-old patient with thalassemia with severely symptomatic heart failure resulting from iron cardiomyopathy, a cardiac T2* of 9 ms (very high cardiac Fe), and an MRI ejection fraction of 15% (normal, >56%). He was treated according the American Heart Association guidelines for iron cardiomyopathy16 with intensive chelation and was asymptomatic within 3 weeks. Three months later, his cardiac T2* was still 9 ms, indicating very high cardiac Fe, but his ejection fraction was normal at 70%. Removing circulating NTBI/LPI resulted in rapid clinical improvement with the same level of tissue iron. Other examples of improved clinical functions before tissue iron is normalized support the notion that the major source of iron toxicity is the circulating NTBI/LPI and the labile cellular ion (reviewed in Coates et al4 and Coates8 ).
This nonlinearity in toxicity complicates clinical management. In general, the clinical manifestations of iron toxicity are silent for many years, perhaps becoming first apparent with pituitary loading and delayed puberty. Furthermore, cardiac loading cannot be predicted from LIC measures. It takes about 10 years for cardiac loading to start, but once iron is in the heart, the rate of loading rapidly increases and can result in significant cardiac iron in a few months. Risk for cardiac dysfunction is clearly related to cardiac T2* and becomes significant with cardiac T2* less than 10 ms.18 At these very high levels of cardiac iron, clinical deterioration can occur in minutes to hours if NTBI/LPI is not controlled. Importantly, cardiac iron toxicity is almost always completely reversible. Endocrine damage is also reversible, but the success is not nearly as predictable.19 Partial adrenal insufficiency is not uncommon in severely iron-loaded patients, and failure to treat when severe stress such as sepsis or acute heart failure are present may have fatal consequences. Last, iron is toxic to the marrow itself. About 45% of patients with low-risk adult myelodysplastic syndrome become transfusion-independent, and more than 60% recover normal levels of white cells and platelets after effective chelation. Similar effects have been seen in other marrow failure syndromes (reviewed in Coates et al4 and Puliyel et al20 ).
Case in point
Our patient had 3 ferritin levels that were quite elevated, yet the LIC was in the low range, showing poor correlation of ferritin and LIC. Nonetheless, there is significant iron in other tissues. The ferritin and LIC have not changed much over the course of 3 years, suggesting his iron input and output are balanced on this dose of chelator (Table 1). His cardiac function remains normal.
Clinically, this indicates that organ function must be monitored along with iron levels. Very importantly, it means that exposure to low levels of reactive iron over decades can cause damage. It is this relation of toxicity to duration of exposure, coupled with the substantially longer survival of patients with chronic transfusion-dependent anemias, that is now giving rise to a new and underappreciated consequence of iron exposure; that is, malignant transformation (reviewed in Coates et al4 and Puliyel et al20 ).
The relative risk for abdominal cancers in patients with thalassemia compared with patients without thalassemia is 1.96 (95% confidence interval [CI], 1.22-3.15; P < .01), and the relative risk for hematological malignancies is 5.32 (95% CI, 2.18-13.0; P < .001).21 Transfused patients with thalassemia have an increased risk for all cancers (hazard ratio, 6.7; 95% CI, 3.29-13.6; P < .001) compared with nontransfused patients with thalassemia. The hazard ratio in transfused patients with thalassemia for abdominal cancer is 9.12 (95% CI, 3.09-27.0; P < .001) and 9.31 for hematological malignancies (95% CI, 1.46-59.3; P < .001) compared with nontransfused patients with thalassemia,21 suggesting a role for iron exposure in cancer risk. There is substantial evidence that iron overload decreases survival in general and increases cancer risk, and that cancer risk is reduced when iron overload is treated (reviewed in Coates et al4 and Puliyel et al20 ). These data motivate our recommendation to aim at maintaining iron levels in a normal range if monitoring resources are available to do so safely.
Principles of iron overload management
The goals of iron overload treatment are to protect tissues from iron-mediated damage, reduce plasma and cytosolic levels of reactive labile iron (Fe+2; NTBI/LPI) to normal, remove all excess iron from the body, and preserve organ function. In our opinion, regardless of the underlying disease process, the major goal of treatment should be to maintain the reactive forms of iron in the normal range throughout life. If this can be accomplished, most of the complications of iron overload can be reduced or even eliminated.4,19,22,23 However, achieving this goal without risking unacceptable treatment toxicity may be difficult, and some experts do not agree with trying to keep iron at near normal levels. These levels cannot be safely achieved using ferritin measures alone as a surrogate for organ iron loading. Thus, it is clear that safely maintaining iron levels in the low range (LIC < 3 mg/g) should only be attempted at centers with the ability to monitor organ iron by MRI and with expertise in chelation.
Diagnosis and monitoring of patients with iron overload
The details of our approach to clinical diagnosis, treatment, and monitoring of iron overload have been recently reviewed elsewhere.24 LIC is the best measure of total body iron loading, as it is linearly related to total body iron content.5 LIC can be measured by MRI imaging, which is more accurate and noninvasive, and has now essentially replaced the use of liver biopsy in developed countries.25 However, significant errors in quantitation can occur if the radiology center is not trained in making these measurements. The MRI scanner produces numbers that are described as T2 and T2* (“T 2 star”), expressed in milliseconds, and decrease in a nonlinear fashion as iron increases. R2 and R2* are the reciprocals of T2 and T2*, respectively, and increase as iron increases.25 A normal LIC is between 0.8 and 1.5 mg/g dry weight of liver. Quantitation by MRI becomes unreliable at LIC greater than 35 mg/g dry weight. Details of MRI techniques have recently been reviewed.25 Most calibrations are performed on a 1.5-Tesla MRI machine, but 3-Tesla MRI machines are becoming more common because of their better image resolution. Nevertheless, magnet strength dramatically alters the iron measurements, and this must be taken into account when comparing measurements over time and between centers.25
Ferritin levels and transferrin saturation are simple tests that have been used for many decades to diagnose and monitor iron overload. Ferritin correlates with total iron in populations of subjects, but can be very misleading in individuals. For example, the trends in ferritin are opposite to the trends in total iron 23% of the time.26 Figure 4 shows serial ferritin and LIC measures in a patient with thalassemia over the course of several years. Ferritin seems to adequately reflect iron content for a while, but then rises even though the total iron is stable at near normal levels. If MRI is not available, ferritin can be used to adjust treatment; however, it is impossible to reduce iron to near normal levels safely using ferritin alone because of the very poor relation to total iron.4,24,27 We rely on LIC measures by MRI for critical decisions about changes in chelation, regardless of ferritin levels, especially when the LIC is less than 3 to 5 mg/g. We do follow ferritin levels for convenience, but because of significant measurement-to-measurement variability, we measure ferritin frequently, usually with each transfusion. Only the ferritin trends should be used for therapeutic decision-making, especially if iron levels are likely approaching a normal range. In general, we recommend getting LIC measurements annually, and sooner if ferritin trends are not consistent with the clinical circumstance.24 Especially at low LIC levels, treatment decisions should be made with great caution when based on ferritin alone.

Time course of iron loading measured by MRI LIC and ferritin in a patient with thalassemia. Ferritin reflects the total iron well during period A but diverges significantly during period B. Ferritin values fluctuate significantly from point to point (from Taher et al,26 with permission).
Time course of iron loading measured by MRI LIC and ferritin in a patient with thalassemia. Ferritin reflects the total iron well during period A but diverges significantly during period B. Ferritin values fluctuate significantly from point to point (from Taher et al,26 with permission).
Transferrin saturation is the only commonly measured test that reflects the toxic NTBI/LPI pool. If the transferrin saturation is greater than 50%, NTBI/LPI levels are likely to be high, and if it is greater than 70%, NTBI/LPI are definitely significantly elevated.28,29 Thus, by inference, the half-life of NTBI/LPI is very short, making transferrin saturation and direct LPI measurements of limited use for monitoring iron chelation therapy.29,30 As the pancreas only loads if NTBI/LPI levels are high and can easily be imaged by MRI at the same time as the liver, we use pancreatic iron as a surrogate for NTBI/LPI area under the curve. If the pancreatic iron level is going up, it suggests that the patient is missing days of chelator, even if the LIC and ferritin are within the desired range.
Cardiac iron must be measured directly by MRI and cannot be predicted on the basis of LIC values. Cardiac T2* of 8 ms or less predicts arrhythmia and heart failure.18 Symptomatic iron cardiomyopathy is life threatening and requires hospitalization and an intensive approach to chelation, using cardiomyopathy guidelines established in patients with thalassemia.16
Pituitary iron, pituitary volume, pancreatic iron, and renal iron can also be measured by MRI, but these measurements are not standard or routinely available.31 We have never observed cardiac iron loading in the absence of pancreatic iron loading, although the converse is not true.32 Thus, if pancreatic iron is not detectable, we believe it is not necessary to assess cardiac iron as frequently.
Treatment of iron overload
Obviously, reducing the amount of transfused blood can help decrease iron loading. However, given the effectiveness of chelation and the consequences of chronic anemia, we do not favor this approach. Transfusion should not be withheld if it is being used to suppress marrow activity (thalassemia, sickle cell anemia, congenital dyserythropoietic anemia). Phlebotomy has been the mainstay for treatment of iron overload when marrow function is normal, but it is not appropriate in the face of ineffective erythropoiesis or marrow failure. Red cell exchange transfusion significantly reduces iron loading in sickle cell anemia,33 but it is not appropriate for patients with thalassemia or marrow failure.
Chelation should start when patients have received 10 transfusions and have a serum ferritin greater than 1000 ng/dl or LIC greater than 3 mg/g dry weight liver. We rarely start chelation in children before 2 years of age because of concerns about theoretical effects of chelation on cognitive development. Furthermore, it is extremely unlikely to have significant clinical iron toxicity by this age.24
Three licensed iron chelators are available in the United States and Europe. Decades of experience have shown that they are effective at reducing iron,34,35 and there is substantial evidence that iron reduction with these agents improves the clinical outcome in thalassemia,2 as well as in MDS.4
All 3 chelators are very effective at controlling iron individually or in combination. The most important differences between these agents relate to their route of administration, half-life, and toxicities. Deferoxamine has a 30-minute half-life and must be given by continuous subcutaneous or intravenous infusion. DFP is an oral chelator and is usually dosed every 8 hours. DFX is also given orally and has the longest half-life of about 14 hours. The major toxicities are renal for DFX, especially at low total iron, and the rare (1.5%) idiosyncratic incidence of agranulocytosis for DFP. Regardless, these medications can be used safely when managed by a clinician experienced in chelation therapy.24 Although all 3 chelators can lower NTBI/LPI and cardiac iron, DFP appears to be the most effective at protecting and restoring cardiac function.16,36
The primary goal of chelation is to clear the circulating reactive forms of iron and to protect tissue from iron toxicity. Clinically, this translates into keeping the NTBI/LPI levels in the normal range, essentially zero, by having circulating chelator present at all times. We use the chelators alone or in combination24,37,38 (off-label use) to maintain NTBI/LPI near zero at all times.24
Another goal of chelation is to eliminate excess total body iron and normalize tissue iron in the heart and endocrine organs. The risk for chelator toxicity is higher at low total body iron (LIC) levels; thus, it is important to monitor renal function very closely and reduce chelation as the LIC approaches the 5 to 3 mg/g levels. Liver enzymes can be 3 to 5 times normal when the LIC is more than 3 to 5 mg/g, but usually resolve as iron levels are controlled; therefore, transaminitis should not prevent starting chelation. It is our practice to reduce chelation when LIC approaches 3 mg/g, and to let the ferritin rise before checking the LIC again. We then increase chelation by a small amount and try to establish the lowest chelator dose that maintains LIC near the 1.5 to 2.5 mg/g range with no toxicity. As long as the patient is transfused, chelation should not be completely stopped, except in case of chelator toxicity.
Case in point.
Our patient had low total iron, but very high tissue iron, when we met him in 2017. We kept his total DFX dose at 22 mg/kg (JadeNu), or about 80% of the maximum dose because of concern about DFX toxicity at this low LIC (Table 1). The daily total dose was divided into twice a day starting in 2017 (off-label use) to provide better NTBI/LPI chelation in the second half of the day. Under this regimen, the LIC came down only slightly over the next 2 years, but the pancreas, pituitary, and cardiac iron levels improved significantly, suggesting much better control of NTBI/LPI.
The presence of organ failure is the primary determinant of treatment intensity and urgency, more so than tissue iron levels. However, iron cardiomyopathy is a special case, and cardiac T2* lower than 10 ms warrants very close follow-up, intensification of treatment, and frank discussion with the patient regarding proper adherence to treatment. At these cardiac iron levels, arrhythmia and heart failure can appear quickly. Other organ failure generally occurs over a period of many months to years. Most often, there is sufficient time for the physician to work with the patient to arrive at an acceptable and effective chelation regimen. Furthermore, as mentioned before, there is some protection from iron toxicity as soon as the chelator is circulating.4,6 For this reason, we try to adjust chelator regimens including combinations of chelator37,38 to maximize circulating chelator levels considering the patient’s lifestyle and tolerance, but recommending chelation 7 days a week as the ideal goal.
Patient adherence to therapy is the main determinant of chelation success
Although there are data showing different absorption rates for some chelators, poor adherence to the recommended therapy is the dominant cause for treatment failure.40 The main goal is to arrive at an effective plan that the patient can closely follow, even if the chosen regimen is not optimal based on pharmacology.
We do not use detailed dosing schedules based on ferritin or LIC, as have been used in some clinical trials. If the LIC is more than 5 to 10 mg/g or the ferritin is more than 2500, we try to maximize chelator doses. When these levels drop below an LIC of 5 mg/g or a ferritin of 1500, we reduce the dosing. Dosing is based on tolerance to the medication, the LIC response, and whether the patient is being regularly transfused or on red cell exchange as we have described.24
Given the marked increased long-term survival and chance of curative treatments in these disorders, preventing organ damage, and reducing risk because of iron at time of transplant, we feel that normalization, at least near-normalization of total iron (LIC < 1.5 to 2.5) and normalization of tissue iron is an ideal goal that should be considered.
Acknowledgments
The authors thank Martine Torres for critical reading and editing of the manuscript.
Correspondence
Thomas D. Coates, Children’s Center for Cancer, Blood Disease and BMT, Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, 4650 Sunset Blvd, MS 54, Los Angeles, CA 90027; e-mail: tcoates@chla.usc.edu.
