
Abstract
Multicenter trials in children and young adults using second-generation CD19-targeted chimeric antigen receptor (CAR) T cells have shown dramatic levels of remission in patients with multiply relapsed/refractory disease (80% to ≥90%). Early results in adult trials have also shown significant responses, and strategies aimed at mitigating toxicities associated with the therapy have improved tolerability. Therefore, if available, CAR T-cell therapy deserves consideration for salvage of children and adults with B-lineage acute lymphoblastic leukemia (B-ALL) who are multiply relapsed, refractory, or relapsed after a previous allogeneic transplantation. For patients with a first relapse or who have persistent minimal residual disease (MRD) after initial or relapse therapy, treatment with blinatumomab or inotuzumab is reasonable to help patients achieve MRD− remission before definitive therapy with allogeneic hematopoietic cell transplantation (HCT). A number of studies in younger patients using 4-1BB–based CAR T-cell constructs lentivirally transduced into patient T cells and then optimally expanded have resulted in long-term persistence without further therapy. In 1 study using CD28-based CARs in adults, the benefit of HCT after CAR T-cell therapy was not clear, because a group of patients experienced long-term remissions without HCT. These data suggest that CAR T-cell therapy may be able to substitute for transplantation in many patients, avoiding the risks and long-term consequences of HCT. With this is mind, and with emerging data better defining ways of enhancing CAR T-cell persistence and avoiding relapse through antigen escape, CAR T cells will have a growing role in treatment of both pediatric and adult B-ALLs in the coming years.
Learning Objectives
Review current results of CD19-targeted CAR T-cell therapy for B-ALL in children and adults, and contrast them with other therapeutic options
Discuss current and upcoming improvements in the process of and approach to CAR T-cell therapy that may allow broader application among B-ALL patients in the near future
Introduction
Although event-free survival (EFS) of children age 1 to 10 years exceeds 80%, infants, teenagers, and adults with B-lineage acute lymphoblastic leukemia (ALL) are at high risk of relapse.1 Risk factors for relapse have been carefully defined. Primary refractory disease, persistence of minimal residual disease (MRD) after initiating therapy for 9 to 12 weeks, and genetic markers have been noted to define very high risk disease for patients receiving initial therapy.2 Any patient relapsing is at very high risk of poor outcome, especially relapses involving marrow that occur within 2 to 3 years of initiating therapy.3 For patients noted to be at very high risk of relapse, hematopoietic cell transplantation (HCT) has offered an opportunity for cure by providing intense therapy and broadly targeted immune therapy through the graft-versus-leukemia effect.4
Although HCT has been shown to improve survival for patients with high-risk ALL, there are significant barriers to success that limit which patients can receive and be cured with HCT. Most importantly, their disease needs to be responsive, because those with even low levels of MRD pre-HCT are at high risk of relapse.5 In addition, myeloablative regimens, especially those containing full-dose total-body irradiation for younger patients, have been noted to improve outcome, and many relapsed or high-risk patients may not meet eligibility requirements for such regimens because of morbidities acquired during therapy.6 These challenges mean that only a fraction of high-risk patients actually undergo HCT, limiting the impact of transplantation as an intervention.
In the past several years, however, highly active immune and cell therapies have revolutionized treatment of high-risk B-ALL. US Food and Drug Administration (FDA) approval of CD19-targeted chimeric antigen receptor (CAR) T cells,7-9 CD19-targeted bispecific T-cell engager molecules,10,11 and CD22-targeted immunotoxins12 has caused clinicians to completely rethink how best to treat high-risk B-ALL patients. Approaches taken by centers vary widely depending upon availability of these agents and experience and preferences of practitioners. This review will focus on the use of CD19-targeted CAR T cells in B-ALL, detailing data from pediatric and adult trials, discussing practical aspects of the treatment, and touching on challenges often experienced. The review will describe CAR T cells in the context of existing immunotherapies, including HCT, followed by a description of novel CAR T-cell approaches just beginning to be tested.
CAR T-cell background/types
The idea of creating a method of forcing T cells to recognize cancer by coupling an antibody-derived single-chain variable fragment (scFv) targeting a tumor-surface protein to the CD3ζ intracellular signaling domain of the T-cell receptor was described nearly 30 years ago.13 Early trials showed limited responses, which were greatly improved through sequential studies, first adding 1 (second generation) and then 2 (third generation) costimulatory domains (CD28 and 4-1BB for second generation and additionally CD27, ICOS, and OX40 for third generation).14 Figure 1 illustrates the CAR protein on a T cell; the main types of CARs discussed in this review contain either CD28 or 4-1BB costimulatory elements coupled with CD3ζ. It is critical to understand that results using CARs may vary dramatically because of the binding affinity of the particular scFV with the target, the hinge length and transmembrane domain, and the intracellular costimulatory and T-cell receptor elements present.
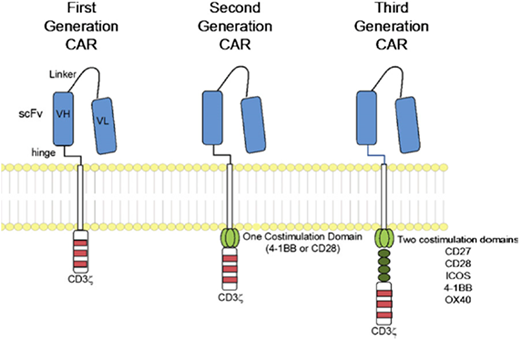
CAR structure, according to signaling domains. CAR molecules link scFv to intracellular signaling domains. The intracellular component includes the CD3ζ alone (first generation) or in combination with 1 (second generation) or 2 (third generation) costimulatory domains. Reprinted with permission.45
CAR structure, according to signaling domains. CAR molecules link scFv to intracellular signaling domains. The intracellular component includes the CD3ζ alone (first generation) or in combination with 1 (second generation) or 2 (third generation) costimulatory domains. Reprinted with permission.45
Published data on anti-CD19 CAR T-cell therapy
Children and YAs
Tables 1 and 2 present data from CAR trials in pediatric and adult patients. Direct comparisons of the trials are difficult, because disease criteria varied, with some trials allowing MRD and others requiring frank relapse (>5% blasts in bone marrow). In addition, approaches to HCT and CRS treatment also varied.
Pediatric and YA trials of CD19-directed CAR T-cell therapy
| Reference | T-cell engager | Population | CR/MRD− CR | Persistence EFS/OS |
|---|---|---|---|---|
| 8 (single center) | CD19 scFv/4-1BB CD3ζ | 25 pediatric/YA patients (age 5-22 y) | 90%/82% (infused patients) | 6-mo EFS, 67% (infused)/OS, 78% (infused) |
| 5 adult patients (age 26-60 y) | 3 patients BMT in CR, 1 patient DLI due to MRD | |||
| 15 | CD19 scFv/ CD28 CD3ζ | 21 pediatric/YA patients (age 5-27 y) | 70%/60% (all enrolled patients) | 9.7-mo OS, 52% |
| LFS of 12 patients achieving MRD− was 78.8%; intent was BMT; 10/12 MRD− proceeded to BMT; 2 ineligible for BMT relapsed | ||||
| 16 | CD19 scFv/4-1BB CD3ζ | 45 pediatric/YA patients (age 1-25 y) | 89% (all enrolled), 93% (infused); all patients with MRD− CR | 1-y EFS, 50.8% (infused)/OS, 65.9% (infused) |
| 11 patients received BMT | ||||
| 9 (ELIANA multicenter trial) | CD19 scFv/4-1BB CD3ζ | 92 enrolled, 75 infused pediatric/YA patients (age 3-23 y) | 66% (all enrolled) | 1-y EFS, 50% (infused)/OS, 76% (infused) |
| 81% (infused); all CRs required 2 time points 4 wk apart and were MRD− | 8 patients BMT (6 in MRD− remission, 2 with MRD+ disease) |
| Reference | T-cell engager | Population | CR/MRD− CR | Persistence EFS/OS |
|---|---|---|---|---|
| 8 (single center) | CD19 scFv/4-1BB CD3ζ | 25 pediatric/YA patients (age 5-22 y) | 90%/82% (infused patients) | 6-mo EFS, 67% (infused)/OS, 78% (infused) |
| 5 adult patients (age 26-60 y) | 3 patients BMT in CR, 1 patient DLI due to MRD | |||
| 15 | CD19 scFv/ CD28 CD3ζ | 21 pediatric/YA patients (age 5-27 y) | 70%/60% (all enrolled patients) | 9.7-mo OS, 52% |
| LFS of 12 patients achieving MRD− was 78.8%; intent was BMT; 10/12 MRD− proceeded to BMT; 2 ineligible for BMT relapsed | ||||
| 16 | CD19 scFv/4-1BB CD3ζ | 45 pediatric/YA patients (age 1-25 y) | 89% (all enrolled), 93% (infused); all patients with MRD− CR | 1-y EFS, 50.8% (infused)/OS, 65.9% (infused) |
| 11 patients received BMT | ||||
| 9 (ELIANA multicenter trial) | CD19 scFv/4-1BB CD3ζ | 92 enrolled, 75 infused pediatric/YA patients (age 3-23 y) | 66% (all enrolled) | 1-y EFS, 50% (infused)/OS, 76% (infused) |
| 81% (infused); all CRs required 2 time points 4 wk apart and were MRD− | 8 patients BMT (6 in MRD− remission, 2 with MRD+ disease) |
BMT, bone marrow transplantation; CR, complete remission; LFS, leukemia-free survival; OS, overall survival; YA, young adult.
Trials of CD19-directed CAR T-cell therapy involving older adults
| Reference | T-cell engager | Population | Response | Persistence/OS |
|---|---|---|---|---|
| 20 | CD19 scFv/4-1BB CD3ζ | 32 enrolled, 30 infused (age 20-73 y; median 40 y) | CR: 91% (all enrolled), 97% (infused); MRD−: 84% (all enrolled), 90% (infused) | Addition of fludarabine increased persistence; 13/30 alive in CR (8 BMT, 5 no BMT); 13/30 BMT (8 alive, 5 died [2 relapse, 3 TRM]) |
| 46 | CD19 scFv/ CD28 CD3ζ | 26 enrolled, 20 infused | 4/5 ALL patients infused (MRD− CR) | 4/5 in CR with median follow-up 5 mo (range, 3-16 mo) |
| Adults (age 20-68 y; ALL median 25 y); cells collected from 13 sibling and 7 unrelated donors; 5 ALL patients | ||||
| 21 | CD19 scFv/4-1BB CD3ζ | 51 total: 42 relapsed/ refractory (age 3-68 y, median 11 y), 9 MRD+ (age 2-44 y, median 24 y) | 86% of RR patients CR/CRi, with 81% MRD− | 27/45 responding patients had BMT; 21/27 alive, median 133 d post-BMT; remainder no BMT; 9/18 relapsed |
| 100% of MRD+ patients became MRD− | ||||
| 22 | Humanized CD19 CAR/4- 1BB CD3-ζ/ T2A-EGFRt | 18 infused | First CAR T-cell infusion: 87% CR, 73% MRD−; 1/3 second CAR T-cell infusions MRD− CR | 6-m OS, 66% |
| 10 pediatric patients (age 3-15 y) | 4 patients underwent BMT (2 allogeneic, 2 autologous) | |||
| 8 adult patients (age 19-57 y) | ||||
| 3 failed murine CAR T-cell therapy | ||||
| 23 | CD19 scFv/4-1BB CD3ζ | Cohorts (median age 44 y, range 21-72 y) | ORR by cohort | Early abstract |
| 1-2: 6 patients, 5 × 107 | 1-2: 33% | |||
| 3: 6 patients, 5 × 108 | 3: 50% (3 died sepsis/CRS) | |||
| 4: 12 patients 5 × 108 (split doses) | 4: 83% (split dosing) | |||
| 24 | CD19 scFv/ CD28 CD3ζ | 83 enrolled, 53 infused | CR: 53% (all enrolled), 83% (infused); MRD−: 39% (all enrolled), 67% (infused) | Median EFS, 6.1 mo; median OS, 12.9 mo |
| Adult patients (age 23-74 y, median 44 y) | If CR and no BMT, 9/26 alive; if CR and BMT received, 5/17 alive |
| Reference | T-cell engager | Population | Response | Persistence/OS |
|---|---|---|---|---|
| 20 | CD19 scFv/4-1BB CD3ζ | 32 enrolled, 30 infused (age 20-73 y; median 40 y) | CR: 91% (all enrolled), 97% (infused); MRD−: 84% (all enrolled), 90% (infused) | Addition of fludarabine increased persistence; 13/30 alive in CR (8 BMT, 5 no BMT); 13/30 BMT (8 alive, 5 died [2 relapse, 3 TRM]) |
| 46 | CD19 scFv/ CD28 CD3ζ | 26 enrolled, 20 infused | 4/5 ALL patients infused (MRD− CR) | 4/5 in CR with median follow-up 5 mo (range, 3-16 mo) |
| Adults (age 20-68 y; ALL median 25 y); cells collected from 13 sibling and 7 unrelated donors; 5 ALL patients | ||||
| 21 | CD19 scFv/4-1BB CD3ζ | 51 total: 42 relapsed/ refractory (age 3-68 y, median 11 y), 9 MRD+ (age 2-44 y, median 24 y) | 86% of RR patients CR/CRi, with 81% MRD− | 27/45 responding patients had BMT; 21/27 alive, median 133 d post-BMT; remainder no BMT; 9/18 relapsed |
| 100% of MRD+ patients became MRD− | ||||
| 22 | Humanized CD19 CAR/4- 1BB CD3-ζ/ T2A-EGFRt | 18 infused | First CAR T-cell infusion: 87% CR, 73% MRD−; 1/3 second CAR T-cell infusions MRD− CR | 6-m OS, 66% |
| 10 pediatric patients (age 3-15 y) | 4 patients underwent BMT (2 allogeneic, 2 autologous) | |||
| 8 adult patients (age 19-57 y) | ||||
| 3 failed murine CAR T-cell therapy | ||||
| 23 | CD19 scFv/4-1BB CD3ζ | Cohorts (median age 44 y, range 21-72 y) | ORR by cohort | Early abstract |
| 1-2: 6 patients, 5 × 107 | 1-2: 33% | |||
| 3: 6 patients, 5 × 108 | 3: 50% (3 died sepsis/CRS) | |||
| 4: 12 patients 5 × 108 (split doses) | 4: 83% (split dosing) | |||
| 24 | CD19 scFv/ CD28 CD3ζ | 83 enrolled, 53 infused | CR: 53% (all enrolled), 83% (infused); MRD−: 39% (all enrolled), 67% (infused) | Median EFS, 6.1 mo; median OS, 12.9 mo |
| Adult patients (age 23-74 y, median 44 y) | If CR and no BMT, 9/26 alive; if CR and BMT received, 5/17 alive |
BMT, bone marrow transplantation; CRS, cytokine release syndrome; ORR, overall response rate; OS, overall survival; TRM, treatment-related mortality.
Initial response
Using a CD19 scFv/4-1BB CD3ζ CAR, investigators at the Children’s Hospital of Philadelphia (CHOP) and the University of Pennsylvania demonstrated remarkable efficacy in children and YAs with B-ALL, with a remission rate of 90% for infused patients who were multiply relapsed or refractory (Table 1).8 National Institutes of Health (NIH) investigators published similar results in children/YAs using a CD19 scFv/CD28 CD3ζ CAR, achieving 70% remission when all enrolled patients were included.15 Using a CD19 scFv/4-1BB CD3ζ CAR with an EGFR surface domain that allows investigators to eliminate the CAR T cells with cetuximab, researchers from Seattle Children’s Hospital followed with a similarly high rate of remission (89% of enrolled patients with MRD− CR).16 These impressive results spawned efforts aimed at rapid regulatory review, resulting in FDA approval of tisagenlecleucel based upon results of the ELIANA trial, run at 25 centers in the United States, Canada, Japan, Australia, and Europe.9 The ELIANA trial required remission to be documented twice within the first 3 months of infusion at least 28 days apart; by this criterion, MRD− remission occurred in 81% of infused patients. The current FDA indication includes children and YAs age 1 to 25 years with multiply relapsed or refractory CD19+ ALL.
Longer-term outcomes
Each trial assessed long-term outcomes differently, and therapies administered in addition to CAR T cells varied significantly (Table 1). Overall survival at 1 year exceeded 50% in all trials. The rate of HCT after CAR T cells varied; HCT was planned as part of the NIH study, but use varied over time with other studies, as investigators became more confident with long-term data.
Older adults
The third review in this educational session is focused on toxicities associated with highly active anti-CD19 CAR T-cell therapies. They are substantial, with 80% to 90% of patients experiencing some degree of CRS and 20% to 40% of patients experiencing severe CRS requiring intensive care unit treatment. In addition, up to 40% of patients experience neurological symptoms,17,18 including rare cases of fatal cerebral edema.19 It is important to keep in mind, however, that a large majority of patients have mild courses, and the experience of patients has been substantially better than that of patients undergoing allogeneic HCT (little to no risk of graft-versus-host disease), especially HCT using myeloablative regimens. Children seem to tolerate CAR T-cell therapy better, with limited data in older adults.
One key issue driving concern is the closure of a Juno Therapeutics–sponsored anti-CD19 CAR T-cell trial in adults as a result of deaths secondary to diffuse cerebral hemorrhage.19 Outside of this specific trial, the incidence of this complication has been rare to absent in other trials. In general, adult patients who proceed to infusion have tolerated early toxicities, and similar high rates of remission have been achieved with low rates of fatal toxicities (Table 2). A report from the Fred Hutchinson Cancer Research Center (FHCRC) adult group showed a 91% CR rate for all enrolled patients, with 90% of infused patients achieving an MRD− remission.20 This phase 1 dose-escalation trial defined the importance of adding fludarabine to cyclophosphamide to achieve improved expansion and long-term persistence/survival. In the 17 adult patients who received fludarabine/cyclophosphamide in the trial, 1-year disease-free survival exceeded 60%. Two Chinese studies including children and older adults used murine21 or humanized22 4-1BB–based CARs. Rates of remission similar to those in pediatric studies are reported, along with longer-term survival exceeding 50%, with a number of patients undergoing HCT. Early data from the University of Pennsylvania study on the 4-1BB–based approach used by the CHOP group showed improved outcomes but increased toxicity with higher doses in adults; 3 patients receiving high doses had fatal toxicities.23 This toxicity issue is confused by the fact that all 3 patients had severe infections complicating their CRS. Additional patients fared better with 2 lower doses, allowing a second dose only if patients did not have significant CRS. This agent is currently being tested in a randomized trial in adults compared with blinatumomab/inotuzumab immunotherapies.
The best documented adult experience is detailed in the most recent publication by the Memorial Sloan Kettering Cancer Center group.24 Of 83 enrolled patients, 53 were infused. This led to 83% achieving CR, with 67% becoming MRD−. This approach included a CD19 scFv/CD28 CD3ζ CAR, which had been noted to have limited persistence25 ; therefore, the group attempted to take eligible patients to allogeneic HCT. Median EFS in their experience was 6.1 months, and median overall survival was 12.9 months.
CAR T cells: bridge to HCT vs curative therapy
Initial studies of highly active second-generation anti-CD19 CAR T-cell approaches were focused on patients who either did not attain remission after initial or relapsed therapy, had multiple relapses, or had relapsed disease after HCT. Long-term survival in such a population is anticipated to be <10%, so achievement of 1- to 2-year survival in more than half of this population is astounding and raises the question of whether this therapy should either replace HCT or be a bridge to facilitate a more efficacious HCT. To address this question, we will take a closer look at the longer-term data available to date.
CD28-based CARs
Both animal and human experience has demonstrated that approaches using second-generation anti-CD19 CD28-based CARs lead to highly active CAR T cells with robust expansion and high likelihood of remission but relatively short persistence (median duration, 14 days [range, 7-138 days] in the Park et al24 study).25 If prolonged presence of CAR T cells is always needed for cure, a transplantation would be necessary after a CD28-based CAR; however, in the Park et al study, if a patient achieved MRD− remission, HCT did not improve survival (however, survival was low with or without HCT, in the 20%-30% range). The most important predictor of outcome in the Park et al study was low disease burden (<5% marrow blasts) at the time of infusion. The combination of low disease burden leading to best outcome is consistent with CD28 CARs leading to major log reduction in responsive patients, sufficient for long-term remission/possible cure in patients starting with low disease burden. The NIH experience with younger patients showed much better outcomes when patients receiving CD28 CARs were able to undergo HCT (of 12 patients achieving MRD− CR, EFS was 79%, with survival of all 10 patients who were eligible for and underwent HCT).15 With this in mind, CD28-based CAR approaches may be better if paired with HCT. Data using xenogeneic mouse models suggest combined CD28, CD3ζ, and 4-1BB third-generation vectors may have prolonged persistence,26 but this has not been verified in human trials.
4-1BB–based CARs
Data from the CHOP single-center experience8 and the ELIANA trial9 show that if younger patients are infused with the 4-1BB–based CAR tisagenlecleucel and achieve remission, >60% will have continued remission at 1 year. Median persistence was 168 days, with many persisting beyond 3 to 5 years. Although data are early, the survival curve after 9 months is flat in both trials (Figure 2). Seattle Children’s Hospital16 and FHCRC adult20 data show similar results, with long-term persistence in a portion of patients who received appropriate lymphodepletion regimens before CAR T-cell infusion. Culture methods for these studies include CD3/CD28 beads, shown to be important in longer-term persistence.27 Culturing processes of the 2 groups differ, but because both approaches result in extended persistence and remission in some patients, these data illustrate that CAR T cells can persist for several years, and persistence leads to long-term remission in a portion of patients.
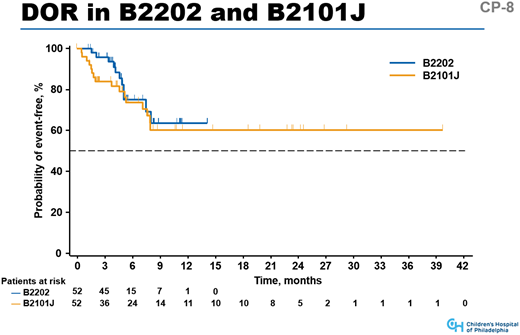
Duration of response (DOR) for patients in the CHOP B2101J trial and ELIANA global registration trial for tisagenlecleucel (B2202). The graphs include patients who achieved CR in both trials; median follow-up was 18.6 months (range, 2.8-43.5 months) in the B2101J trial and 9.3 months in the B2202 trial (range, 3.1-18.5 months).
Duration of response (DOR) for patients in the CHOP B2101J trial and ELIANA global registration trial for tisagenlecleucel (B2202). The graphs include patients who achieved CR in both trials; median follow-up was 18.6 months (range, 2.8-43.5 months) in the B2101J trial and 9.3 months in the B2202 trial (range, 3.1-18.5 months).
Challenges to CAR T-cell efficacy
There are 3 biological challenges that have led to failure in a portion of patients treated with anti-CD19 CAR T-cell therapy. The first is lack of initial expansion of collected lymphocytes in culture; the second, loss of CART T cells early in therapy; and the third, antigen escape.
Initial expansion of infused CAR T cells
Many studies have sought to optimize culture conditions for CAR T cells. Early studies showed that CD3/CD28 beads led to better expansion and persistence,27 whereas others showed a benefit to performing separate CD4 and CD8 cultures and delivering the cells to recipients at a fixed ratio (Figure 2).28 However, culture conditions are only part of the story, because even under the best of conditions, 5% to 10% of cultures from patients do not expand. The quality of the lymphocytes entering culture was explored by investigators at CHOP. After demonstrating that early-lineage T cells were essential for expansion,29 the group went on to report the efficacy of lymphocyte expansion under CAR T-cell culture conditions in 157 pediatric patients with a number of cancers before and after receiving several types of lymphotoxic chemotherapy.30 They noted lower efficacy of expansion with tumor types other than ALL (solid tumors and lymphoma), suggesting an effect of tumor type on lymphocytes. They also noted worsening efficacy after chemotherapy treatment, especially in children age <3 years. Prior use of cyclophosphamide and doxorubicin was especially harmful to expansion. Patients with low numbers of naïve T cells and cells reliant on glycolysis were at risk of failure, whereas use of fatty acids such as palmitate improved the expansion ability of T cells. These data suggest improved outcomes may be associated with early apheresis procedures in patients slated for CAR T cells, preferably before initiation of salvage chemotherapy. The data also suggest that development of approaches that result in ideal T-cell profiles would be useful.
Early loss of CAR T cells
Loss of CAR T cells to date has been associated with either T cell–intrinsic issues preventing expansion or T cell–extrinsic immunological rejection. In a study of patients receiving tisagenlecleucel for chronic lymphocytic leukemia, investigators noted that patients who did not respond had T cells expressing genes associated with effector differentiation, glycolosis, exhaustion, and apoptosis, whereas expansion and response were associated with the presence of CD27+PD1−CD8+ CAR T cells with high levels of interleukin-6 receptors (Figure 3).31 Long-term persistence of CAR T cells was associated with the presence of increased numbers of CD24+CD45RO−CD8+ T cells in the collection before expansion.
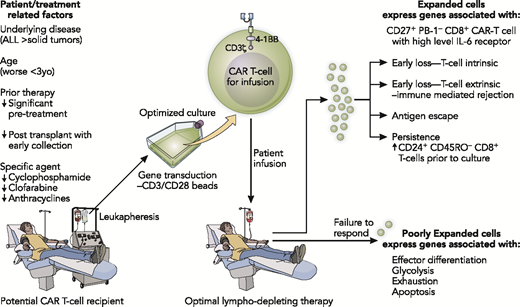
Key biological variables that can affect outcome through the CAR T-cell collection, expansion, and infusion process. Illustration by Patrick Lane, ScEYEnce Studios.
Key biological variables that can affect outcome through the CAR T-cell collection, expansion, and infusion process. Illustration by Patrick Lane, ScEYEnce Studios.
Another important cause of early CAR T-cell loss extrinsic to the T cell is likely immunological rejection, but this has been difficult to characterize. Studies with tisagenlecleucel have examined the presence of antibodies against CAR T cells, either present before or stimulated by CAR T-cell treatment, and have found no correlation with response or persistence.32 Investigators at the FHCRC noted T cell–mediated rejection in 5 patients with very early CAR T-cell loss,20 so some rejection could be T cell mediated, but to date, rejection has not been well characterized.
Antigen escape
One pattern of relapse after CAR T cell–induced remission follows very early loss of CAR T cells or loss of B-cell aplasia (absolute B cells measured by flow rising above 50 to 100/μL) and has generally been associated with CD19+ B-cell relapses. When CAR T-cell remissions are MRD− and cells persist, relapses are more commonly CD19−.9 Studies have revealed that CD19 is still present on the cells, but various splice variants that have deleted the binding site for the CAR have been described.33 Studies of CD22-targeted CARs are under way, with early reports of remissions in more than half of patients.34 Because these trials generally enroll patients for whom CD19-targeted approaches have failed, comparisons between efficacy using this target potentially do not represent outcomes that would have been achieved if this target had been used as an upfront approach. Combined antigen approaches to avoid antigen escape are reviewed in the Future directions in CAR T cell therapy for ALL section and Figure 4.
Treatment approaches: CAR T cells vs other immune therapies
Targeted engagers/immunotoxins
Blinatumomab is a bispecific T-cell engager that works by binding the CD3+ receptor on T cells with CD19. This has resulted in significant activity, including CR in 30% to 40% of relapsed/refractory pediatric or adult patients presenting with frank relapse.10,11 In patients who are responsive to chemotherapy, however, and are in a CR with MRD, approximately 70% to 80% can become MRD−.35 Although a fraction of patients have experienced longer-term remission with continued therapy, generally HCT has been used when treatment with curative intent is planned and patients are eligible. Therefore, the ideal setting for this agent is in patients with MRD+ disease who have a donor available for a first HCT. Although neurotoxicity, CRS, and tumor lysis can occur, the safety profile of blinatumomab is better than intensive chemotherapy.
Inotuzumab ozogamicin, an anti-CD22 antibody-toxin conjugate, has been more effective than blinatumomab for achieving remission when patients have frank relapse (CR rates nearing 60%), but it has a disadvantage in that it is clearly associated with venoocclusive disease/sinusoidal obstruction syndrome, especially if followed by allogeneic HCT.12 To mitigate this challenge in patients subsequently undergoing HCT, consensus recommendations are published36 that include limiting inotuzumab to 2 courses, avoiding dual alkylator preparative regimens, and avoiding combinations of hepatotoxic agents during HCT. Adding rituximab in combination with chemotherapy for patients with CD20+ B-ALL has been shown to be beneficial for initial therapy,37 but the role of targeting CD20 in refractory B-ALL is not well established.
Case for CAR T cells: refractory, multiply relapsed, or relapsed after HCT
Despite the risk of high-grade CRS in a portion of patients and the rare risk of life-threatening neurotoxicity, because CD19-targeted CAR T-cell therapy has a 70% to 90% success rate in attaining an MRD− remission, treatment with CAR T cells should strongly be considered if commercial or research approaches are available to a relapsed/refractory patient. It is best to pursue this therapy at the point of relapse, rather than trying and failing a series of other approaches. Because a majority of patients with full marrow relapse will not attain remission with blinatumomab, CARs may offer a better chance at remission. Inotuzumab is also challenging because of the need to proceed to HCT for cure and because of the increased VOD risk,38 although it is an important option if a CD19− clone is present. If a patient has relapsed after an allogeneic HCT, because treatment with CD19-targeted 4-1BB–based CAR T cells can lead to prolonged remission without the need for a second transplantation in up to half of patients, this therapy should be considered the treatment of choice.
First relapse or patients in CR with MRD
With a first relapse, reinduction with chemotherapy is generally recommended, although a recent study showed a survival benefit with blinatumomab compared with chemotherapy,39 and promising responses with inotuzumab in combination with chemotherapy have been seen.38 If a patient attain remissions and is MRD−, a suitable donor for HCT is present, and the patient is eligible for HCT, allogeneic transplantation is appropriate. However, if a patient remains MRD+ after reinduction, treatment with either CAR T cells, blinatumomab, or inotuzumab is worth considering, because continued intensive chemotherapy courses are sequentially less likely to put patients into an MRD− remission,40 and further intense therapy increases the likelihood of organ damage or infection that could limit HCT options.
Optimizing CAR T-cell therapy
Early referral and collection
Because commercial tisagenlecleucil is under a risk evaluation mitigation strategy, it is currently limited to regional sites. Many sites have research protocols available. The key to optimizing therapy for patients if CAR T-cell therapy is not available is establishing a referral relationship with a convenient site. As outlined previously, collection of T cells that are not exhausted because of intense chemotherapy may help improve outcomes. Many CAR T-cell centers are able to arrange for early collection, and collection before salvage chemotherapy is worth pursuing. For commercial tisagenlecleucel, collection can occur at the administering centers before insurance approval for manufacturing, so in this situation, insurance approval need not impede early collection. If disease control before collection is required, clinicians should avoid highly lymphotoxic chemotherapies such as clofarabine.
Interim therapy after collection
Once collection has been completed, the center performing CAR T-cell therapy may have recommendations for interval therapy, but in general, the key is to avoid toxic reinduction regimens. Although disease control is important, remission is not required, so intermediate dose regimens or maintenance-type approaches are best if disease control can be maintained.
After CAR T-cell therapy, transplantation or not?
Some CAR T-cell approaches, such as CD28-based vectors or current universal off-the-shelf CAR T-cell approaches are well documented to have short periods of persistence, generally 4 to 8 weeks. In these situations, moving to HCT may be beneficial if patients have a donor available and are eligible. However, should patients receiving 4-1BB–based approaches proceed to transplantation once they are stable in remission after CAR T-cell infusion? Early loss of detectable CAR T cells increases risk. Notably, however, patients often have CAR T cells go below the level of detection and maintain anti-CD19 CAR T-cell activity shown by persistent B-cell aplasia. Longer-term studies have shown an association between persistence of B-cell aplasia and decreased risk of relapse (although CD19− relapse can still occur).41 Persistence of B-cell aplasia for 9 to 12 months seems to result in a lower risk of relapse; however, the number of patients in longer-term follow-up is limited.
Unfortunately, we cannot yet reliably predict which patients will have CAR T-cell persistence. In addition, patients always run a risk of CD19− relapse. With that in mind, it is reasonable to offer HCT to individuals who are in an MRD− remission after CD19-targeted CAR T-cell therapy if they have a donor and are eligible. However, with >60% of young patients who achieve an MRD− remission after tisagenlecleucel having persistence at 1 year and with relapse after that time point being less common, many individuals may choose to risk CD19− relapse in the hope of a long-term remission without HCT. If patients have already undergone allogeneic HCT before CAR T-cell therapy, second or third HCT is associated with high risks of treatment-related mortality, and it may be best to move to HCT only after loss of B-cell aplasia. Loss of B-cell aplasia within 3 to 6 months carries a high risk of relapse, and second infusions of CAR T cells in this group have generally not been sustained, so a donor for HCT should be identified so such patients can move quickly to HCT if eligible. Some patients who lose CAR T cells/B-cell aplasia late (after 6 months) may have prolonged presence of CAR T cells and B-cell aplasia after second infusions, so this can be considered as an alternative approach to HCT. For most patients who lose CAR T cells after 9 to 12 months, relapse is rare, and a second infusion likely to succeed in attaining remission, so it is reasonable to watch and wait.
Future directions in CAR T cell therapy for ALL
Moving CAR T cells earlier into therapy
As CAR T-cell therapies become more effective and safer, it is easy to envision moving this strategy earlier into therapy to avoid short- and long-term chemotherapy complications. However, with the toxicity profile of tisagenleceucel and related agents in mind, currently the most reasonable strategy of moving earlier into therapy consists of targeting high-risk patients who would otherwise undergo HCT and attempt to cure a portion of patients with CAR T cells alone, reserving HCT as a rescue strategy. The Children’s Oncology Group is embarking on such a study aimed at children with persistent detectable MRD after consolidation (9-12 weeks into therapy); 5-year EFS in these children is poor,42 and the goal of the trial is to improve EFS, while avoiding HCT in at least 50% of the patients treated.
Antigen escape and flexible targeting
CD19 loss is better termed escape, because studies have shown that resistance is due to splice variants in the persistent CD19 protein that remove the loci engaged by CAR T-cell scFv.33 This could be circumvented by different targets, the most well studied of which is CD22. Early experiences with CD22-targeted CARs have shown CR in up to 70% of patients34 ; however, resistance can develop quickly, most likely from downregulation of CD22 surface expression. Better approaches to combat resistance will likely involve targeting multiple antigens,43 and a number of centers are assessing CD19/CD22 dual-targeted regimens. Because of the likelihood of resistance to any give antigen, multitargeted approaches make sense. There are a number of approaches (Figure 4): targeting 2 antigens with the same construct, targeting 2 antigens with separate constructs in a single cell, or targeting antigens with pools of cells with 1 or the other construct. Which of these approaches would best address resistance is unclear, but results of multiple antigen–targeting CAR studies will be coming forward soon.

Methods of avoiding CAR T-cell escape (A-C) or limiting CAR T-cell targeting to allow use of multiple antigens for either cell specificity or avoidance (D-E): targeting 2 antigens with the same construct (A), targeting 2 antigens with separate constructs in a single cell (B), targeting antigens with pools of cells containing the 1 or the other construct (C), positive gating strategy (2 antigens on the tumor required for killing) (D), and negative gating strategy (a second antigen present on a normal cell shuts down killing) (E). Illustration by Patrick Lane, ScEYEnce Studios.
Methods of avoiding CAR T-cell escape (A-C) or limiting CAR T-cell targeting to allow use of multiple antigens for either cell specificity or avoidance (D-E): targeting 2 antigens with the same construct (A), targeting 2 antigens with separate constructs in a single cell (B), targeting antigens with pools of cells containing the 1 or the other construct (C), positive gating strategy (2 antigens on the tumor required for killing) (D), and negative gating strategy (a second antigen present on a normal cell shuts down killing) (E). Illustration by Patrick Lane, ScEYEnce Studios.
To broaden the application of CAR T cells to other leukemias and solid tumors, multiple targeting will likely be necessary, because clear single-antigen targets are not obvious (Figure 4). Other strategies that assist in targeting CAR T cells to tumors include use of a second antigen that either confirms cancer or is associated with a normal cell and shuts down a CAR reaction. In addition, microenvironment issues that shut down CAR T cells, including PD-1 and other cytokines, will need to be overcome by approaches using either systemic or CAR T cell–secreted molecules.44
CAR T-cell persistence
Persistence of CAR T cells has been associated with a favorable naïve T-cell phenotype present during collection and higher levels of expansion. Use of checkpoint inhibitors is also being studied, as well CAR constructs designed to secrete various cytokines,44 both aimed at improving persistence or overcoming regulatory environments to improve effect. These studies may allow the creation of a CAR T-cell approach that is more consistent, predictable, and controlled.
Turning off CAR T cells
A number of centers have put kill switches of various sorts into their constructs.16,20 These have acted as safety measures for excessive toxicities, but in the near future, such switches may be useful in shutting down CAR T cells that have served their purpose. Although likely to vary with disease type, there is growing evidence that CD19-targeted 4-1BB approaches that persist for 9 to 12 months lead to very low chances of relapse, even if CAR T cells later become undetectable and B-cell aplasia is eventually lost. It is conceivable that, as with chronic myeloid leukemia patients going off tyrosine kinase inhibitors, we may be able to turn CAR T cells off and restore B-cell function in a portion of patients. Such studies are important for moving anti-CD19 CAR T-cell approaches into more general use in B-ALL, especially in children, because the prospect of the necessity of extended or lifelong therapy with IVIG impedes thoughts of making this treatment acceptable frontline therapy for lower-risk disease.
Conclusion
CAR T cells are a potent form of cell therapy that offers a better chance of attaining remission in relapsed/refractory adult and pediatric B-ALL than other recently approved immune therapies, and a portion of patients can be cured without further therapy. Although the toxicity profile is high, the procedure is safer than allogeneic HCT, containing little to no risk of graft-versus-host disease. Efforts aimed at avoiding antigen escape and enhancing persistence, along with earlier initiation of therapies for CRS, will continue to improve outcomes. Patients considering use of CAR T-cell therapy may benefit from early T-cell collection and avoidance of highly toxic chemotherapy approaches while awaiting cell manufacturing.
Correspondence
Michael A. Pulsipher, Children’s Hospital Los Angeles, 4650 Sunset Blvd, Mailstop #62, Los Angeles, CA 90027; e-mail: mpulsipher@chla.usc.edu.
