
Abstract
Eosinophilia is associated with a wide variety of allergic, rheumatologic, infectious, neoplastic, and rare idiopathic disorders. Clinical manifestations range from benign asymptomatic presentations to life-threatening complications, including endomyocardial fibrosis and thromboembolism. The prognosis and choice of treatment depend not only on the degree of eosinophilia and severity of organ involvement, but also on the etiology of the eosinophilia. Unfortunately, despite recent advances in molecular and immunologic techniques, the etiology remains unproven in the overwhelming majority of cases. This review presents a practical approach to the diagnosis and treatment of patients presenting with unexplained marked eosinophilia. A brief overview of the mechanisms of eosinophilia and eosinophil pathogenesis is also provided.
Learning Objectives
Describe the spectrum of eosinophilic disorders, from benign eosinophilia to eosinophilic leukemia
Discuss the mechanisms of eosinophil-associated pathogenesis and general approaches to treatment
Eosinophils are terminally differentiated cells of the myeloid lineage characterized by the presence of secondary granules that stain pink with the acidic dye, eosin.
First described in 1900 by Paul Ehrlich, eosinophils have long been regarded as inflammatory cells that play an important role in protection against helminth infection and the pathogenesis of allergic inflammation. More recently, with the advent of murine models devoid of eosinophils and novel therapies that target eosinophils in humans, a clearer understanding of the function of eosinophils in health and disease is beginning to emerge. This has important implications with respect to the diagnosis and treatment of eosinophilic disorders.
Regulation of eosinophilia
The balance between eosinophil production, trafficking from the bone marrow to the tissues, and apoptosis is a crucial determinant of blood and tissue eosinophilia.
Regulation of eosinophilopoiesis from CD34+ stem cells in the bone marrow is dependent on complex interactions between key transcription factors, including GATA-1, CCAAT/enhancer-binding protein, and PU.1,1 and on the presence of eosinophil-promoting cytokines, the most important of which is interleukin (IL)-5.2,3 Eosinophilopoiesis also occurs in extramedullary tissues, particularly in the setting of allergic inflammation, where it is driven to a large extent by IL-5 secreted by tissue resident type 2 innate lymphoid cells.4 Consistent with a central role of IL-5 in eosinophilopoiesis, surface expression of IL-5 receptor α on the human common myeloid progenitor is an essential and early step in eosinophil lineage commitment and is preserved even in neoplastic eosinophils.5 Other cytokines, including IL-2, IL-3, and GM-CSF, also promote eosinophilopoiesis and appear to be relatively more important in some clinical scenarios, such as adenocarcinoma-associated eosinophilia. The intrinsic mechanisms by which eosinophilopoiesis is downregulated are less well understood, and murine models suggest that there are checks and balances at many different levels. Examples include the eosinophil surface receptor, paired immunoglobulin-like receptor A,6 and microRNA-21.7
Whereas the majority (>90%) of eosinophils are found in the tissues in normal individuals, their distribution is relatively restricted to the gastrointestinal tract, spleen, lymph nodes, thymus, mammary glands, and uterus. In eosinophilic disorders, migration of increased numbers of eosinophils to these and other organs occurs in response to local production of mediators, including IL-5, the eotaxin chemokines (eotaxin/CCL11, eotaxin-2/CCL24, and eotaxin-3/CCL26), CCL5/RANTES, and non-chemokine factors, such as complement factor C5a, platelet-activating factor (PAF) and leukotrienes.8 These mediators are produced by a wide variety of cells, including lymphocytes, mast cells, epithelial cells, and eosinophils themselves. The mechanisms by which eosinophils traffic preferentially to specific organs in different eosinophilic disorders is poorly understood and remains an active area of research.
Finally, increased production of many of the same cytokines involved in eosinophilopoiesis, including IL-5, IL-3 and GM-CSF, has been implicated in the prevention of eosinophil apoptosis in the blood and tissues.9 This is likely counterbalanced, at least in part, by engagement of eosinophil inhibitory receptors, such as Siglec-8, which induces eosinophil death upon binding to endogenous glycans in inflamed tissues.10
Eosinophils and pathogenesis
Eosinophils have been implicated in the pathogenesis of tissue fibrosis,11 thrombosis,12 vasculitis,13 and allergic inflammation. The propensity of eosinophils to cause these effects depends on a number of factors, including the number of eosinophils, their location, and degree of activation. Although these factors may be influenced by the underlying etiology of the eosinophilia, the consequences of eosinophilic inflammation can be identical despite markedly different clinical diagnoses. For example, clinically indistinguishable eosinophilic endomyocardial fibrosis has been described in patients with PDGFRA-positive myeloproliferative neoplasm, idiopathic hypereosinophilic syndrome, and parasitic helminth infection.
Activated eosinophils contribute to disease pathogenesis both through direct cytotoxic effects and by recruitment and activation of other inflammatory cells. Tissue deposition of eosinophil granule proteins (major basic protein, eosinophil-derived neurotoxin, eosinophil cationic protein, and eosinophil peroxidase) contained in the characteristic secondary granules of eosinophils plays a major role in direct tissue damage. Granule proteins can be released from intact eosinophils through a process called piecemeal degranulation, whereby selective secretion of individual granule components occurs without disruption of the cell membrane, or from “cell-free” granules liberated by exocytosis or during extracellular DNA trap cell death (ETosis).14,15 In addition to the granule proteins, a wide array of cytokines and chemokines are stored preformed in the secondary granules and can be secreted in response to specific signals, leading to the recruitment and activation of other cells involved in the inflammatory response, including lymphocytes, mast cells, and fibroblasts.16,17 Eosinophil activation also leads to secretion of reactive oxygen intermediates and the formation of increased numbers of lipid bodies, the primary site of synthesis of eicosanoids, inflammatory mediators that include leukotriene C4 and 5-lipoxygenase.18
Spectrum of hypereosinophilic syndromes
The definition of hypereosinophilic syndromes (HESs) has been a subject of controversy since 1968 when Hardy and Anderson first used this term to describe 3 patients with marked peripheral eosinophilia and cardiopulmonary manifestations.19 In fact, when Chusid et al published their landmark series of patients with HES in 1984, they recognized that “there is a continuum of hypereosinophilic disease with eosinophilic leukemia at one pole” and included patients with clear evidence of an eosinophilic myeloproliferative neoplasm as well as patients with more benign phenotypes of HES.20 Patients with secondary treatable causes of eosinophilic disease, such as parasitic infections, were excluded. Using this definition, the clinical manifestations of HES are extremely varied, ranging from relatively asymptomatic disease to endomyocardial fibrosis and thromboembolism. In a large multicenter study of patients with HES, the most common organ systems involved at presentation were, in order of descending frequency, skin, lung, and gastrointestinal tract.21 Although <5% of patients had cardiac and neurologic involvement at presentation, these complications ultimately developed in 20% of patients, highlighting the importance of early recognition and effective treatment in these disorders.
Recent advances in molecular and immunologic techniques have enabled more definitive etiologic classification in some cases of HES, most notably those associated with eosinophilic myeloproliferative neoplasms or aberrant lymphocyte populations. This, together with the increasing availability of targeted therapies, including tyrosine kinase inhibitors and monoclonal antibodies, has dramatically improved prognosis in HES, but has further complicated the terminology as groups of experts struggle with the best way to define and classify patients with eosinophilic disorders of known and unknown etiologies in a way that facilitates treatment decisions.22-24 For a review, see Klion.25 Although much attention has focused on blood eosinophil levels, marked blood eosinophilia can be present in the absence of clinical symptoms,26 and tissue eosinophilia can cause significant clinical manifestations in the absence of peripheral eosinophilia.27 Furthermore, as mentioned above, the clinical manifestations of eosinophilia can be identical in eosinophilic disorders of very varied etiologies (Figure 1).

Frequency distribution of diagnoses in a cohort of 302 subjects referred for evaluation of unexplained hypereosinophilia. Adapted from Klion.25
Frequency distribution of diagnoses in a cohort of 302 subjects referred for evaluation of unexplained hypereosinophilia. Adapted from Klion.25
For the purposes of this review, hypereosinophilic syndrome (HES) will be defined in the broadest sense as: (1) hypereosinophilia (HE; a peripheral eosinophil count >1.5 × 109/mL documented on at least 2 occasions) or marked tissue eosinophilia, and (2) clinical manifestations directly attributable to the eosinophilia or presumed to be due to eosinophilia and for which no alternative cause can be identified. This definition captures all patients with clinical manifestations due to eosinophilia regardless of the underlying etiology, including those due to primary abnormalities involving the eosinophil lineage and those driven by cytokines and other mediators secreted by lymphocytes or other cells, whether due to intrinsic abnormalities in these cells or driven by external stimuli, such as parasite antigens or environmental allergens.
The advantage of defining HES in this way is that it provides a starting point for a pragmatic approach to any patient presenting with eosinophilic disease. The challenge is the classification of individual patients into meaningful categories that assist in the selection of the most appropriate treatment. To this end, the following six classification categories have been proposed25 (Figure 1; Table 1): (1) myeloproliferative HE/HES (M-HE or M-HES), including both definite and presumed eosinophilic myeloproliferative neoplasms (MPN); (2) lymphocytic variant HE/HES (L-HE or L-HES), in which an aberrant or clonal lymphocyte population drives eosinophilia through the production of soluble mediators; (3) overlap HES or eosinophilic disease restricted to a single organ system; (4) associated HE/HES or HE/HES in the setting of a distinct diagnosis (ie, parasitic helminth infection, drug hypersensitivity and primary immunodeficiency) in which eosinophilia has been described in a subset of affected patients; (5) familial HE/HES, a rare autosomal dominant disorder; and (6) idiopathic HES. Hypereosinophilia of unknown significance (HEUS) refers to untreated HE in the absence of clinical symptoms, evidence of a myeloproliferative neoplasm or treatable secondary cause.26 HEUS may progress to HES in some cases.28 This classification system both differs from and overlaps with the 2008 WHO guidelines,23 which group myeloid and lymphoid neoplasms, including those characterized by marked eosinophilia, on the basis of a variety of genetic, histopathologic, and clinical criteria.
Classification of hypereosinophilic syndromes
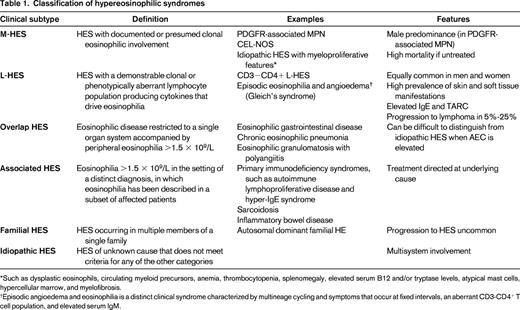
*Such as dysplastic eosinophils, circulating myeloid precursors, anemia, thrombocytopenia, splenomegaly, elevated serum B12 and/or tryptase levels, atypical mast cells, hypercellular marrow, and myelofibrosis.
†Episodic angioedema and eosinophilia is a distinct clinical syndrome characterized by multineage cycling and symptoms that occur at fixed intervals, an aberrant CD3-CD4+ T cell population, and elevated serum IgM.
Treatment of HES
When confronted with a patient with unexplained eosinophilia, the first question to be addressed is whether immediate therapy is necessary to prevent potentially irreversible, eosinophil-mediated end organ damage. Examples of findings that warrant urgent therapy include clinical evidence of cardiac, neurologic, or thromboembolic complications and/or the presence of extremely high (>100 × 109/L) or rapidly increasing eosinophil counts. Corticosteroids (1 mg/kg to 1 g methylprednisolone depending on the severity of clinical manifestations) are the initial treatment of choice except in situations where an underlying etiology is identified and known to be corticosteroid-resistant. Ivermectin (200 mcg/kg/d × 2 days) should be administered concomitantly to all patients with a history of potential exposure to the helminth, Strongyloides stercoralis, to prevent the potentially fatal corticosteroid-induced hyperinfection syndrome. Although every effort should be made to obtain appropriate diagnostic studies (Table 2) before initiating corticosteroid therapy, treatment should not be delayed for this purpose if the clinical situation is deteriorating. If there is no reduction in eosinophil count after 1-2 days of high dose corticosteroid therapy, a second agent should be selected using the clinical presentation and most likely underlying etiology as a guide (see below).
Initial evaluation of HES

*Substantially affected by corticosteroid therapy.
Adapted from Klion.25
Conventional agents for the treatment of HES include corticosteroids, hydroxyurea, interferon α, and imatinib (the only agent FDA-approved for treatment of HES).21 Numerous additional agents, including chlorambucil, vincristine, etoposide, cladribine, cytarabine, methotrexate, cyclosporine, alemtuzumab, and cyclophosphamide, have been used to treat small numbers of steroid-unresponsive patients with varied success and may be appropriate in some situations. Finally, renewed interest in the role of eosinophils in allergic and inflammatory disorders has led to the development of a wide variety of biologic therapies that directly or indirectly target eosinophils.29
Because systemic corticosteroids are appropriate initial therapy for most patients with HES, identification of subgroups of patients for whom therapies other than corticosteroids are preferable as initial therapy is an essential first step in any treatment algorithm. These include patients with secondary causes that require specific therapy targeting the underlying cause (associated HE/HES), patients with known imatinib-sensitive mutations (such as PDGFR-associated MPN), patients with single-organ involvement that may be responsive to topical corticosteroid therapy (overlap HES) and patients with familial eosinophilia or HEUS, who may not require any therapy at all. The remaining patients with presumed myeloproliferative neoplasms (M-HES), lymphocytic variant HES (L-HES), idiopathic HES, and some patients with overlap HES (those with severe single organ manifestations or evidence of vasculitis) should be treated with systemic corticosteroids. Dosing and duration should be individualized based on the clinical manifestations, comorbidities and perceived risk of serious end-organ damage. Once the eosinophil count has normalized and symptoms have improved, the corticosteroid dose should be tapered slowly. A second agent should be considered in patients who cannot be maintained on 10 mg prednisone equivalent or less daily, or who experience significant side effects.
The choice of a second-line agent depends on a variety of factors, including the clinical and/or molecular variant of HES, site(s) of organ involvement, side effect profile of the drug, and patient preference. The most commonly used second line agents for the treatment of idiopathic HES are hydroxyurea (1-2 g orally daily) and interferon-α (1-3 mU subcutaneously daily), each of which is effective in ∼30% of patients.21 Although it is beyond the scope of this review to provide a guide to therapy for all patients with HES who fail first line therapy, certain situations deserve mention (a more complete approach can be found by Klion25 ).
Myeloproliferative HES (M-HES).
Approximately 20% of patients who present with HES have features suggestive of a myeloproliferative disorder, including the presence of dysplastic eosinophils and eosinophil precursors in the blood, anemia and/or thrombocytopenia, elevated serum B12 and/or tryptase levels, splenomegaly, and bone marrow hypercellularity and fibrosis. The majority of these patients (as many as 80%) have a PDGFRA-associated MPN. As mentioned above, these patients are exquisitely sensitive to imatinib mesylate and should receive treatment with this agent as first-line therapy.30,31 Starting doses of 100-400 mg daily are typically sufficient to induce hematologic remission within days, and molecular remission is almost universal. Patients with evidence of cardiac involvement, based on echocardiographic findings and serum troponin levels, should receive high dose corticosteroids (≥1 mg/kg prednisone or equivalent) during the first few days of therapy to reduce the risk of myocardial necrosis.32 Although resistance to imatinib is uncommon in PDGFRA-associated MPN, it has been reported.30,33 Newer tyrosine kinase inhibitors (TKIs), including sorafenib, midostaurin, and ponatinib, which have in vitro activity against cells carrying the FIP1L1-PDGFRA fusion with imatinib-resistant mutations,34 should be considered in such cases.
Some PDGFRA-negative patients presenting with HES and myeloproliferative features have documented cytogenetic or molecular abnormalities, such as rearrangements involving PDGFRB, FGFR1, or JAK2 or point mutations in KIT. In these cases, treatment should be guided by the underlying abnormality. Other patients have no detectable cytogenetic or molecular abnormalities and do not meet WHO criteria for acute leukemia or CEL-NOS. Although high-dose corticosteroids are often effective in the short-term reduction of eosinophilia and clinical manifestations in these patients with “idiopathic” M-HES and are useful in initial management, transient or partial responses are common. Given the possibility of occult imatinib-sensitive mutations and the clinical similarity between patients with idiopathic M-HES and those with PDGFRA-associated disease, imatinib would seem a logical second-line agent in steroid-resistant patients. In fact, recent data from our center suggests that myeloproliferative features are an important predictor of imatinib response in steroid-resistant, PDGFRA-negative subjects with HES. Alternative therapies for patients who do not respond to steroids or imatinib include hydroxyurea, interferon-α, second and third-generation tyrosine kinase inhibitors, and allogeneic transplantation.
Lymphocytic variant HES (L-HES).
L-HES is defined by the presence of clonal and/or aberrant lymphocytes that produce soluble mediators that drive eosinophilia. Patients with L-HES often present with skin and soft tissue manifestations, elevated serum IgE and thymus and activation regulated chemokine (TARC) levels.35,36 Although most patients with L-HES respond to corticosteroid therapy, many require moderate to high doses. Interferon-α is often used as the preferred second-line therapy due to its dual effect on eosinophils and T lymphocytes. Other agents, including methotrexate, cyclophosphamide, cyclosporine, and alemtuzumab have also been used as steroid-sparing agents in L-HES with variable success. Progression to lymphoma occurs in 5%-25% of patients with L-HES, often many years after the initial presentation,35-37 and may be heralded by increasing or new lymphadenopathy, expansion of the aberrant T-cell population, development of cytogenetic abnormalities, especially 6p-, and/or progressive resistance to treatment.
Novel biologic agents
Despite the dramatic increase in novel biologics in clinical trials for asthma and atopic disease (for review, see Radonjic-Hoesli et al 29 , and Fulkerson and Rothenberg40 ), data in HES is sparse. Following promising pilot trials using two different monoclonal antibodies to IL-5, reslizumab, and mepolizumab, to treat patients with treatment-refractory HES, a double-blind, placebo-controlled trial in 85 PDGFRA-negative patients demonstrated that monthly mepolizumab was safe and effective as a steroid-sparing agent in HES, including L-HES.38 Long-term safety and efficacy of mepolizumab for the treatment of HES was confirmed in an open-label extension study.39 Mepolizumab is available on a compassionate use clinical protocol for patients with life-threatening HES refractory to standard therapies (http://www.clinicaltrials.gov). A clinical trial of benralizumab, an afucosylated monoclonal antibody to IL-5 receptor that has shown efficacy in the treatment of eosinophilic asthma, is also underway in patients with HES. Additional novel targeted therapies directed at inhibitory receptors, eosinophilic chemokines and their receptors, and other molecules involved in the regulation of eosinophil development, activation, migration to tissues, and apoptosis, are under development.29,40 These and other targeted therapies not only provide hope for patients with HES, but are certain to help further define the role(s) of eosinophils in human health and disease.
Acknowledgments
This work was funded by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Correspondence
Amy D. Klion, Human Eosinophil Section, Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 4, Room B1-29, 4 Center Drive, Bethesda, MD 20892; Phone: 301-435-8903; Fax: 301-451-2029; e-mail: aklion@nih.gov.
