
Abstract
Chronic lymphocytic leukemia (CLL) is usually diagnosed in early stage, asymptomatic patients, and, although a wealth of prognostic parameters have been identified, the standard approach is a “watch and wait” strategy irrespective of risk factors. Therapy is only indicated if “active disease” criteria (International Workshop on Chronic Lymphocytic Leukemia guidelines) are met, and the routine upfront treatment is a combination of CD20 antibody (rituximab, ofatumumab or obinutuzumab) and chemotherapy (fludarabine /cyclophosphamide, bendamustine, chlorambucil), with the choice mainly determined by physical fitness of the patient. The major subgroup in which this approach does not result into satisfactory efficacy is in CLL with 17p deletion (17p−) or TP53 mutation (TP53mut). Likewise, patients with a short initial response duration (i.e., <24-26 months) have a dismal outcome with chemoimmunotherapy salvage. Therefore, these patients have been referred to as “ultra high risk,” and, in these subgroups, novel agents such as signaling kinase inhibitors (also termed B-cell receptor signaling inhibitors; e.g., ibrutinib targeting Bruton tryosine kinase, idelalisib targeting phosphoinositide 3-kinase) and BCL2 antagonists (venetoclax, formerly ABT-199/GDC-0199) have shown dramatic efficacy. Ibrutinib and idelalisib are currently approved for the treatment of relapsed or refractory CLL or frontline treatment of 17p−/TP53mut CLL regardless of fitness. Therefore, these agents are challenging the concept of adjusting treatment to fitness and TP53 status, because they offer remarkable efficacy combined with exceptional tolerability. Nevertheless, it appears that 17p−/TP53mut retains an adverse prognostic impact, making additional improvement a primary research goal aimed at the development of the best combinations and/or sequences of these new agents, as well as prognostic and predictive markers guiding their use.
Learning Objectives
“Watch and wait” is the standard of care for early stage, asymptomatic chronic lymphocytic leukemia (CLL) patients irrespective of prognostic factors, but these may inform counseling and follow-up
17p−/TP53mut, physical fitness (age), and duration of previous response are the most critical determinants of treatment choice
Combination of CD20 antibody and chemotherapy, with the choice determined by fitness, is the standard upfront treatment for CLL patients without 17p−/TP53mut
Based on dramatic efficacy and favorable toxicity profile of novel agents such as ibrutinib, idelalisib, and venetoclax, these agents are the preferred treatment approach for “ultra high-risk” CLL (17p−/TP53mut and refractory/early relapse) patients
The role of allogenic stem cell transplantation needs reassessment, and individualized counseling is required for young “ultra high-risk” patients
There is a strong need for additional research on improved treatment and prognostic, as well as predictive markers in the era of novel “biological” approaches, aimed at finite treatment duration and eventually a cure
Standard approach to early stage, asymptomatic patients
Chronic lymphocytic leukemia (CLL) is the most prevalent leukemia in adults, and ∼90% of patients today are initially diagnosed with asymptomatic, early stage (Rai 0-I, Binet A) CLL.1-3 A considerable number of these patients have indolent disease and may have a normal life expectancy, whereas others show rapid disease progression and have poor outcome. Clinical trials evaluating immediate chemo(immuno)therapy in early stage CLL so far failed to show an overall survival (OS) benefit; thus, watch and wait has remained the standard of care.4,5 However, today novel treatment approaches are available (see below), and a wealth of prognostic factors have been identified, including lymphocyte doubling time (LDT), bone marrow infiltration pattern, serum thymidine kinase (TK) and β2-microglobulin (β2-MG) levels, genomic aberrations assessed by fluorescence in situ hybridization (FISH), ZAP70, CD38, and CD49d expression, as well as the mutational status and structure of the immunoglobulin heavy-chain variable genes (IGHV) and gene mutations, most prominently TP53 (for overviews, see Stilgenbauer and Zenz6 and Baumann et al7 ). Numerous single markers and various models, scores, or nomograms have been reported to be associated with prognosis in early stage CLL, but there is a lack of prospective validation within clinical trials (8-14 ). In the CLL1 trial of the German CLL Study Group (GCLLSG), a comprehensive set of markers were evaluated for their prognostic effect, and 17p−, 11q−, unmutated IGHV, β2-MG >3.5 mg/dL, LDT <12 months, and age >60 years were found to be of independent effect for OS (Table 1).15 However, although none of these markers are recommended in current guidelines for management of early stage CLL patients, they are determined frequently in routine practice with no clear rationale for their use. In particular, a small proportion (∼5%) of early stage CLL patients have 17p−/TP53mut CLL,15,16 the “ultra high-risk” marker in chemo(immuno)therapy studies.17-22 Moreover, there is a proportion of early stage patients with indolent disease course despite having 17p−/TP53mut CLL,16,23 and these patients may be characterized by mutated IGHV status, early stage (Rai 0) disease, younger age, good performance status, and normal lactate dehydrogenase. Therefore, the presence of 17p−/TP53mut at diagnosis in an otherwise asymptomatic patient without treatment indication (no “active disease”)1 should not in itself prompt early treatment in routine practice. However, these patients deserve more extensive prognostic workup and closer monitoring (Figure 1).
Factors of independent prognostic effect on outcome in multivariable analysis of OS in the CLL1 trial of the GCLLSG for early stage (Binet A) CLL (n = 534)
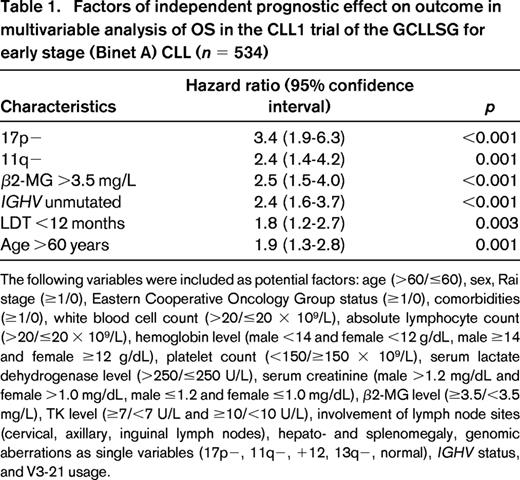
The following variables were included as potential factors: age (>60/≤60), sex, Rai stage (≥1/0), Eastern Cooperative Oncology Group status (≥1/0), comorbidities (≥1/0), white blood cell count (>20/≤20 × 109/L), absolute lymphocyte count (>20/≤20 × 109/L), hemoglobin level (male <14 and female <12 g/dL, male ≥14 and female ≥12 g/dL), platelet count (<150/≥150 × 109/L), serum lactate dehydrogenase level (>250/≤250 U/L), serum creatinine (male >1.2 mg/dL and female >1.0 mg/dL, male ≤1.2 and female ≤1.0 mg/dL), β2-MG level (≥3.5/<3.5 mg/L), TK level (≥7/<7 U/L and ≥10/<10 U/L), involvement of lymph node sites (cervical, axillary, inguinal lymph nodes), hepato- and splenomegaly, genomic aberrations as single variables (17p−, 11q−, +12, 13q−, normal), IGHV status, and V3-21 usage.
![Figure 1. Suggested CLL management algorithm. Assessment [disease biology, patient fitness (age), and treatment history] is indicated in white boxes, and corresponding treatment is in orange boxes. (A) Initial treatment of patients with CLL requires determination of iwCLL criteria for “active disease.” These patients should then be clinically evaluated for fitness and require FISH analysis for 17p13 deletion (17p−) and mutation analysis of TP53. (B) In patients with relapsed/refractory CLL, the response to previous treatment should also be considered; in case of response duration of 2 to 3 years or longer (late relapse), retreatment with a chemoimmunotherapy regimen should be considered. In contrast, patients with response duration of <2 to 3 years should be considered to be treated with nonchemotherapy regimens. FISH analysis for 17p13 deletion (17p−) and mutation analysis of TP53 should be repeated before treatment initiation. Alem indicates alemtuzumab; B, bendamustine; BSC, best supportive care; CR, complete response; Ibr, ibrutinib; Idela, idelalisib; Obi, obinutuzumab; Ofa ofatumumab; PD progressive disease; PR, partial response; SD, stable disease; w & w, watch and wait. Alemtuzumab is no longer licensed for CLL and should be considered only in exceptional cases when other treatments are not an option. Treatment options in frail (very old) patients need highly individualized decisions. (Modified based on the Onkopedia Guidelines of the German Society of Hematology and Oncology, available at www.onkopedia-guidelines.info.)](/view-large/figure/6512139/bep0011504990001.jpeg)
Suggested CLL management algorithm. Assessment [disease biology, patient fitness (age), and treatment history] is indicated in white boxes, and corresponding treatment is in orange boxes. (A) Initial treatment of patients with CLL requires determination of iwCLL criteria for “active disease.” These patients should then be clinically evaluated for fitness and require FISH analysis for 17p13 deletion (17p−) and mutation analysis of TP53. (B) In patients with relapsed/refractory CLL, the response to previous treatment should also be considered; in case of response duration of 2 to 3 years or longer (late relapse), retreatment with a chemoimmunotherapy regimen should be considered. In contrast, patients with response duration of <2 to 3 years should be considered to be treated with nonchemotherapy regimens. FISH analysis for 17p13 deletion (17p−) and mutation analysis of TP53 should be repeated before treatment initiation. Alem indicates alemtuzumab; B, bendamustine; BSC, best supportive care; CR, complete response; Ibr, ibrutinib; Idela, idelalisib; Obi, obinutuzumab; Ofa ofatumumab; PD progressive disease; PR, partial response; SD, stable disease; w & w, watch and wait. Alemtuzumab is no longer licensed for CLL and should be considered only in exceptional cases when other treatments are not an option. Treatment options in frail (very old) patients need highly individualized decisions. (Modified based on the Onkopedia Guidelines of the German Society of Hematology and Oncology, available at www.onkopedia-guidelines.info.)
Suggested CLL management algorithm. Assessment [disease biology, patient fitness (age), and treatment history] is indicated in white boxes, and corresponding treatment is in orange boxes. (A) Initial treatment of patients with CLL requires determination of iwCLL criteria for “active disease.” These patients should then be clinically evaluated for fitness and require FISH analysis for 17p13 deletion (17p−) and mutation analysis of TP53. (B) In patients with relapsed/refractory CLL, the response to previous treatment should also be considered; in case of response duration of 2 to 3 years or longer (late relapse), retreatment with a chemoimmunotherapy regimen should be considered. In contrast, patients with response duration of <2 to 3 years should be considered to be treated with nonchemotherapy regimens. FISH analysis for 17p13 deletion (17p−) and mutation analysis of TP53 should be repeated before treatment initiation. Alem indicates alemtuzumab; B, bendamustine; BSC, best supportive care; CR, complete response; Ibr, ibrutinib; Idela, idelalisib; Obi, obinutuzumab; Ofa ofatumumab; PD progressive disease; PR, partial response; SD, stable disease; w & w, watch and wait. Alemtuzumab is no longer licensed for CLL and should be considered only in exceptional cases when other treatments are not an option. Treatment options in frail (very old) patients need highly individualized decisions. (Modified based on the Onkopedia Guidelines of the German Society of Hematology and Oncology, available at www.onkopedia-guidelines.info.)
In conclusion, watch and wait remains the standard of care for early stage, asymptomatic CLL patients irrespective of risk factors. Nevertheless, the management schedule (e.g., intervals of visits) may be adjusted to the risk profile, i.e., with 3-month follow-up for patients with high-risk markers (e.g., 17p−, 11q−, TP53mut, unmutated IGHV, high β2-MG, and short LDT), but therapeutic decisions based on these markers remain a clinical research question. In particular, there is a strong need for prospective trials evaluating prognostic markers and novel therapies (see below) in early stage CLL, because this is not only the most frequently encountered situation in general practice but may also offer the best chance for long-term disease control, change of the natural disease course, or even cure.
Advanced stage, symptomatic patients with treatment indication (“active disease”)
Conventional chemoimmunotherapy
The current upfront standard of care in physically fit patients with CLL in need of treatment as defined by International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria (“active disease”) is fludarabine, cyclophosphamide, and rituximab (FCR).19,24 This is based on high response rates [40%-50% complete response (CR)], prolongation of progression-free survival (PFS), and notably OS compared with FC. Of note, there is benefit of FCR in almost all genetic patient subgroups (except for patients with 17p−/TP53mut; see below), with particularly long-term remissions in IGHV mutated/genetically favorable CLL.22 However, a large proportion of CLL patients cannot tolerate FCR because of age (e.g., >65-70 years) and comorbidity [e.g., cumulative illness rating score (CIRS) >6, decreased renal function, e.g., creatinine clearance <30-50 mL/min].19,24 Therefore, age and, probably better, fitness (e.g., determined by renal function or scores such as CIRS) are key prognostic determinants for treatment decisions.
The GCLLSG CLL10 study compared FCR with bendamustine plus rituximab (BR) among fit (CIRS ≤6, creatinine clearance ≥70 mL/min) patients without 17p− and showed that overall BR was inferior to FCR in terms of response rate and PFS.25 However, FCR was associated with markedly increased toxicity and only marginally (nonsignificant) superior efficacy in the group of patients over the age of 65 years. Therefore, the conclusion was that BR is a valid alternative treatment option among physically fit CLL patients above the age of 65 years and without 17p−/TP53mut.
For unfit patients, deemed inappropriate for FCR-type treatment, two novel antibodies have been approved. Ofatumumab was licensed in combination with bendamustine or chlorambucil (Clb) based on the randomized phase 3 COMPLEMENT-1 trial that compared Clb with ofatumumab-Clb and showed significantly higher efficacy with only moderately increased toxicity of the combination.26 The phase 3 CLL11 trial compared in a 3-arm design Clb, rituximab-Clb (R-Clb), and obinutuzumab-Clb. Obinutuzumab-Clb showed higher efficacy not only compared with Clb but also with R-Clb with acceptable safety profile, which led to approval of obinutuzumab-Clb for the upfront treatment of unfit patients.27
Therefore, combination of CD20 antibody (rituximab, ofatumumab, or obinutuzumab) and chemotherapy (FC, bendamustine, Clb) is the standard upfront treatment for the vast majority of CLL patients, with the specific choice of agents mostly determined by fitness.
Initial therapy for 17p−/TP53mut CLL
The major exception to the standard of frontline chemoimmunotherapy are patients with 17p−/TP53mut CLL, who have markedly inferior outcomes with chemo(immuno)therapy.17-22 Importantly, this poor efficacy appears to be independent of the IGHV mutation status, because also patients with 17p− CLL and mutated IGHV had a dismal outcome in the CLL8 trial (Figure 2).

PFS of 17p− CLL patients from the CLL8 trial (both treatment arms combined) defined by unmutated (n = 40; green) and mutated (n = 11; blue) IGHV status (median of 11.0 versus 16.2 months, p = 0.359).
PFS of 17p− CLL patients from the CLL8 trial (both treatment arms combined) defined by unmutated (n = 40; green) and mutated (n = 11; blue) IGHV status (median of 11.0 versus 16.2 months, p = 0.359).
The molecular surrogate of 17p− is deletion of the TP53 gene, and the sole mutation of TP53 also has been associated with poor outcome after chemo(immunotherapy) treatment in CLL. In the British CLL4 trial, TP53mut has been identified as adverse prognostic marker after treatment with Clb, fludarabine, and FC.21 In the GCLLSG CLL8 trial, TP53mut and 17p− were the two independent markers of strongest influence on PFS and OS in multivariable analysis after FC and FCR treatment.22 Therefore, 17p− and TP53mut are today grouped together because of the similar biological meaning and clinical consequences.
Other markers, such as 11q− and unmutated IGHV, mutation of other genes (e.g., NOTCH1, SF3B1, and BIRC3), expression of CD38, ZAP70, and CD49d, high β2-MG, and high TK have been associated with inferior outcome in the chemo(immuno)therapy setting.17-27 Nevertheless, these markers do not distinguish a subgroup of CLL patients that requires a different therapeutic approach in general practice and are therefore not used to define a particular risk group, in contrast to 17p−/TP53mut that alters treatment choice. Of note, evaluation of these and other novel prognostic or predictive markers in clinical trials is required before these can be used in general practice. For example, the dramatic prognostic effect of BIRC3 mutation/deletion20 could not be confirmed in a clinical trial setting (see below).
Although the results of FCR are in general superior to those reported for fludarabine or FC, they are still dismal in the 17p−/TP53mut subgroup. For example, in the subset of patients with 17p− CLL enrolled in the randomized CLL8 study comparing FC and FCR, FCR was slightly superior in terms of PFS (median of 11.3 months for FCR versus 6.5 months for FC, p = 0.02), but this was markedly inferior to all other genomic subgroups.19,22,24 Outside of the CLL8 study, the experience with frontline FCR in patients with 17p− CLL has been in general unsatisfactory, because reports from the MD Anderson/Mayo clinic,23 Spanish Group for CLL,28 and the Memorial Sloan-Kettering Cancer Center29 all showed that only ∼25% of patients with 17p− CLL achieve remissions of ≥3 years after frontline FCR or similar regimens.
In line with this, the GCLLSG CLL2M phase 2 study of frontline BR enrolled only 8 patients with 17p− CLL, of which 3 responded; all responses were partial, and the median PFS was only 7.9 months.30 Therefore, BR is likely inferior to FCR as induction therapy of 17p− CLL.
The anti-CD52 antibody alemtuzumab showed efficacy data in refractory CLL that were similar in patients with and without 17p−, whereas other markers, such as β2-MG, retained their prognostic effect.31,32 Pettitt et al33 demonstrated that the combination of alemtuzumab and high-dose corticosteroids was feasible and active in 17p− CLL in the U.K. CLL206 study, the CR rate was very high at 65%, but remissions were only moderately durable, with a median PFS of 18 months. However, because of the inferior risk/benefit ratio compared with novel agents (see below) and its market withdrawal for CLL treatment, there is very little room for alemtuzumab in the treatment of CLL, except for exceptional cases.
Novel agents targeting signaling kinases (e.g., B-cell receptor) pathway and BCL2 in the management of 17p−/TP53mut and relapsed/refractory CLL
Two major classes of novel agents have emerged with substantial activity across all genomic subgroups of CLL, including 17p−/TP53mut, the signaling kinase inhibitors [targeting, for example, the B-cell receptor (BCR) pathway] and the BCL2 antagonists.34,35 These drugs are orally bioavailable and show dramatic efficacy combined with very favorable tolerability compared with chemoimmunotherapy. Biologically, these agents, in contrast to chemo(immune)therapy, do not exert their mode of action through genotoxic mechanisms and should therefore be active irrespective of dysfunctional p53 (i.e., 17p−/TP53mut). However, there are increasing signals that resistance to novel agents may be related to the genomic instability of the CLL clone, and a common pattern of resistance to novel agents is the development of Richter's transformation36-40 (see below). These observations are important because they suggest that selected patients with 17p−/TP53mut CLL may still benefit from allogenic stem cell transplantation (allo-SCT), even in the novel agent era (see above).
Signaling kinase inhibitors (such as ibrutinib and idelalisib)
The BCR pathway is an important survival stimulus in CLL, and a number of small-molecule inhibitors of pathway components have become available in the clinic.34,41-49 These agents typically cause evasion of CLL cells from the tissue compartment into the blood, thus causing a paradoxical, transient, and “benign” lymphocytosis that must be distinguished from disease progression. This condition is termed “partial response with lymphocytosis” (PRL), which may persist over many months and needs to be added to the response criteria.
Most of the data with signaling kinase inhibitors are available with the Bruton tryosine kinase (BTK) inhibitor ibrutinib (PCI-32765) and the phosphoinositide 3-kinase δ (PI3Kδ) inhibitor idelalisib (GS-1101).42-49 Both drugs demonstrate high activity and achieve durable remissions in patients with relapsed/refractory, genetically unselected CLL. Specific to patients with relapsed/refractory 17p− CLL, >200 patients have been treated with ibrutinib monotherapy across 3 clinical trials.42,47,50 These included RESONATE-17, the largest interventional study conducted to date in 17p− CLL.50 In aggregate, these studies showed that ibrutinib monotherapy achieved objective responses (including PRL) in ≥80% of patients with relapsed/refractory 17p− CLL, with PFS of ∼80% at 12 months and projected to be 50%-60% at 24 months. These results compare very favorably in historical comparison even with upfront treatment of 17p− CLL with FCR with a median PFS of <12 months.19,22 However, complete remissions were rare (<5%), and in one study, patients with 17p− CLL had a statistically significant increase in relapse rates and decreased OS compared with patients without 17p− CLL.36
With regard to idelalisib, the monotherapy results in 13 patients with heavily pretreated CLL was less encouraging, with an overall response rate (ORR) of 54% and a median PFS of only 3 months.44 However, the experience of idelalisib in combination with rituximab in a subsequent phase 3 study was substantially better at ORR of 82% and median PFS of 17 months.45,46 Of note, the efficacy of idelalisib plus rituximab appeared not to be affected by the presence of 17p−/TP53mut in a preliminary, retrospective subgroup analysis of this study,51 indicating this combination as another valid treatment option for this subgroup. However, as for ibrutinib, more mature data and systematic comparison of the efficacy of idelalisib in biological CLL subgroups are needed before treatment recommendations in general practice can be derived.
Although the BCR antagonists very rarely achieve complete remissions in the relapsed/refractory setting, the results may be different when these drugs are used in the frontline. For example, 2 CRs were recorded in the 4 treatment-naive subjects with 17p− CLL enrolled on the ibrutinib plus rituximab study,52 and in a study of frontline idelalisib and rituximab, ORR and CR of 100% and 33%, respectively, were reported in 9 patients with 17p− CLL.48 In the largest study evaluating ibrutinib in treatment-naive 17p−/TP53mut CLL, 32 of the 33 evaluable patients achieved a response with OS of 84% at 24 months.49 Notably, both the Food and Drug Administration and the European Medicines Agency have granted approval of ibrutinib and idelalisib plus rituximab for the frontline treatment of patients with 17p−/TP53mut CLL who are inappropriate candidates for chemoimmunotherapy based on the tremendous efficacy of these agents in these patients, despite the lack of randomized comparison against chemoimmunotherapy.
Open issues in the context of signaling kinase inhibitor treatment: response assessment, prognostic markers, and resistance
The distinct mode of action and clinical characteristics of signaling kinase inhibitors have required a reevaluation of several key aspects of general CLL management practice. First, the initial lymphocytosis seen with these agents would result in patients being classified as having progressive disease based on iwCLL criteria, although all other parameters indicate improvement. Of note, prolonged lymphocytosis has been associated with favorable outcome in one study,53 underlining the conceptually different approach with these agents and effect on response assessment including minimal residual disease (MRD; see below).
Second, the novel agents are given as continuous therapy and maximal response is often slowly evolving over time. Thus, when comparing signaling kinase inhibitors with chemo(immuno)therapy in clinical trials, timing of the assessment becomes very important, and arguably, the effect of surrogate endpoints, such as PR, CR, and MRD, on PFS and OS need to be reassessed. In particular, the effect of MRD on PFS and OS needs to be reevaluated because the signaling kinase inhibitors are associated with long-term CLL persistence in blood and marrow despite effective disease control and iwCLL guideline-compatible remission. The mode of continues therapy also leads to serious issues regarding cost and accessibility of novel agents, the discussion of which are beyond the scope of this review, but clearly indicate the need for the development of novel approaches with finite treatment duration.
Third, the role of prognostic markers, such as 17p−, 11q−, unmutated IGHV, TP53mut, and elevated β2-MG, will need to be studied in clinical trials because it appears that outcome is not invariably linked to these as it was with chemoimmunotherapy. This raises questions regarding treatment planning, including consideration of the timing of allo-SCT for young ultra high-risk patients (see below). Although they are historically much superior to any other treatment option, it appears in the most mature analysis that 17p− and 11q− still have negative effects on outcome with ibrutinib treatment.36 Conversely, it appeared in early analyses that, in contrast to chemoimmunotherapy, patients with unmutated IGHV had a better outcome,42 but this was not confirmed at a more mature stage36 and in other trials.49 The likely explanation is that IGHV unmutated CLL shows a more rapid response compared with IGHV mutated CLL, possibly because of a stronger “addiction” to BCR signaling in IGHV unmutated CLL.
Fourth, it is becoming clear that also with signaling kinase inhibitors (and BCL2 antagonists) resistance and disease progression are observed. There are two major patterns of relapse on targeted agent treatment in CLL: (1) early progression mainly attributable to Richter transformation; and (2) late progression mainly attributable to CLL relapse as a result of the acquisition of resistance mutations. 17p−, 11q−, and complex karyotype (all in fact related to each other) have been associated with emergence of CLL progression, and the mechanism of this appears to be the acquisition and clonal selection of BTK (at the ibrutinib binding site cysteine 481) and PLCg2 (the immediate next downstream signaling molecule) mutations, which activate the pathway despite the presence of ibrutinib36-39 (see also the chapter by Jennifer Woyach in this issue). The other prominent feature of treatment failure is the occurrence of Richter transformation, the molecular pathomechanisms of which remain to be determined.
Together, there is preliminary evidence that 11q− and 17p− may retain their prognostic role even in the era of signaling kinase inhibitors, but clearly the reassessment of the role of prognostic markers, the methods of response assessment, and the mechanisms of resistance development are research priorities.
BCL2 antagonists (such as venetoclax/ABT-199/GDC-0199)
The antiapoptotic members of the BCL2 family are targeted by BH3 mimetics (e.g., ABT-737, ABT-263, and ABT-199).35,40,54,55 The current drug in clinical development for CLL is ABT-199 (also known as GDC-0199, Venetoclax), which is specific for BCL2 and bypasses the issue of BCLXL-related thrombocytopenia.35 The results of ABT-199 in CLL have been presented in preliminary form.35,40,55 Both as a single agent and in combination with rituximab, ABT-199 achieved high response rates of ∼80% and importantly—and differently from the results of BCR antagonists—CR rates of ∼25% were reported even in the relapsed/refractory 17p− setting. A number of patients have become MRD negative after ABT-199 (with or without rituximab) therapy, and anecdotal evidence suggests that ABT-199 may be discontinued in remission without recurrent disease at early follow-up.40,55 A pivotal phase 2 study of ABT-199 in 17p− CLL (frontline and relapsed/refractory) is currently underway. Efficacy of ABT-199 has been observed across all genomic subgroups, including 17p−/TP53mut CLL, although there is a continuous slow pattern of relapse, making the identification of prognostic markers and biology of resistance urgent scientific questions.
allo-SCT
There is unequivocal evidence that allo-SCT is effective in CLL and may be associated with prolonged disease-free survival even in patients with advanced, chemotherapy-refractory disease and in 17p−/TP53mut CLL.56-61 Of note, allo-SCT is the only therapeutic procedure where 17p-/TP53mut CLL appears to have outcomes not different from those of all other genetic subgroups, i.e., 17p−/TP53mut loses its prognostic effect.58-61 Approximately 40% to 50% of ultra high-risk patients remain in long-term remission and are potentially cured after allo-SCT, but the substantial morbidity and mortality (still ∼20%) put restrictions on its use. Prognostic factors associated with inferior transplant outcomes included pretreatment with >3 lines of therapies, advanced clinical stage, marked lymphadenopathy, and refractory disease at the time of transplant.58-61 Thus, allo-SCT should be performed in 17p−/TP53mut and relapsed/refractory CLL while therapeutic options for remission induction still exist and preferably in the absence of extensive tumor burden. In the chemoimmunotherapy era, consolidation with allo-SCT would have been considered for all fit CLL patients with early (within 24-36 months) relapsed or refractory disease, as well as 17p−/TP53mut in the upfront setting.6,56 This position is now questioned in view of the promising results of the novel agents (see above) in CLL, and a recent ERIC/EBMT consensus paper weighing the risk/benefit profile was coming to the conclusion that there is little room for allo-SCT in the upfront setting.62 Even in early relapse and for patients with 17p−/TP53mut CLL, there is no universal indication for allo-SCT, and a careful consideration of the risk profile needs to be performed including genetics (17p−/TP53mut, 11q−), age, comorbidities, and degree of donor matching. Nevertheless, and to facilitate this, immediate referral of patients with early relapse and/or 17p−/TP53mut CLL to a transplant center for counseling is recommended. Of note, a very recent study indicated that salvage after SCT failure can be achieved with conventional chemoimmunotherapy and novel agents, stressing the need for additional research on the integration of allo-SCT and novel agents in a holistic treatment concept for ultra high-risk CLL.63,64
Additional developments and new concepts
New genomics: next-generation sequencing and novel gene mutations
The prognostic value of other pathogenic CLL “driver gene” mutations in the definition of “high-risk” disease and the management of patients with CLL is less well defined. There are data from heterogeneous cohorts outside clinical trials that mutation of genes, such as NOTCH1, SF3B1, and BIRC3, could improve the prognostic accuracy of genomic analysis.20 However, clinical trials, such as U.K. CLL4 and GCLLSG CLL8 have only partly confirmed an independent prognostic role of these gene mutations, and their effect on outcome was much less pronounced then 17p−/TP53mut.21,22 A provocative facet that needs additional study is the potential predictive influence of NOTCH1mut for a reduced benefit from the addition of rituximab to FC, i.e., as a truly predictive marker guiding treatment choice.22
On a global genomic scale, the presence of subclonal driver mutations as detected by whole exome sequencing (WES) has been linked to inferior outcome,65 and this association was confirmed in a recent subgroup analysis from the CLL8 trial66 : the presence of both a pretreatment subclonal driver (66.5% of subjects) and clonal driver (84.2%) was associated with significantly shorter PFS [hazard ratio (HR) of 1.64, p = 0.003 and HR of 1.78, p = 0.009], with only subclonal drivers achieving significance in the FCR arm. In this study, cases with mutated TP53 and SF3B1 had significantly shorter PFS and OS, whereas XPO1 (HR of 2.02, p =0.02), BRAF (HR of 2.05, p = 0.05), and EGR2 (HR of 2.14, p = 0.05) mutations were associated with reduced OS and trend toward shorter PFS, but ATM and BIRC3 (Figure 3) mutations had no significant effect on outcome, highlighting the need of more clinical trial data to establish new prognostic markers. From a pathogenesis and disease evolution perspective, this analysis also identified a temporal relationship and evolution of acquired genetic variants.66 Surprisingly, genomic aberrations, such as 11q−, +12, and 13q−, appeared to precede gene mutations, such as ATM, BIRC3, and FBXW7. In turn, analysis of matched pretreatment and relapse samples reveal patterns of clonal evolution in relation to chemo(immune)therapy, pointing to resistance-“driving” variants with increasing allelic frequency compared with “passengers” with varying or decreasing ratio (Figure 4).

PFS of CLL patients from the CLL8 trial (both treatment arms combined) defined by the presence (n = 12; blue) and absence (n = 266; orange) of BIRC3 mutation.66
PFS of CLL patients from the CLL8 trial (both treatment arms combined) defined by the presence (n = 12; blue) and absence (n = 266; orange) of BIRC3 mutation.66

Clonal architecture and evolution as determined by mathematical calculation and analysis of sequential samples by WES from the CLL8 trial.66 (A) Temporal directed sequence of acquisition of genomic variants inferred from the observation of driver events found at clonal and subclonal frequency in the same samples. Distinct points of origin restricted to a few events, such as 13q− and +12, with early convergence to 11q− and subsequent divergence in diverse late-occurring driver mutations. (B) Comparison of the cancer cell fractions (CCFs) of the putative driver alterations between baseline and relapse samples showed clonal shifts in 57 of 59 cases, of which 21 showed linear and 36 branched evolution with the latter significantly associated with FCR treatment. Notably, variants inferred as early events by temporal network were stably clonal over time. Late events demonstrated either increasing (i.e., in TP53) or shifting (i.e., in SF3B1, ATM) CCFs, suggesting evolution in relation to therapy (grey = stable, red = increasing, and blue = decreasing allele fraction).
Clonal architecture and evolution as determined by mathematical calculation and analysis of sequential samples by WES from the CLL8 trial.66 (A) Temporal directed sequence of acquisition of genomic variants inferred from the observation of driver events found at clonal and subclonal frequency in the same samples. Distinct points of origin restricted to a few events, such as 13q− and +12, with early convergence to 11q− and subsequent divergence in diverse late-occurring driver mutations. (B) Comparison of the cancer cell fractions (CCFs) of the putative driver alterations between baseline and relapse samples showed clonal shifts in 57 of 59 cases, of which 21 showed linear and 36 branched evolution with the latter significantly associated with FCR treatment. Notably, variants inferred as early events by temporal network were stably clonal over time. Late events demonstrated either increasing (i.e., in TP53) or shifting (i.e., in SF3B1, ATM) CCFs, suggesting evolution in relation to therapy (grey = stable, red = increasing, and blue = decreasing allele fraction).
BCR “stereotypes,” antigenic drive, and autonomous signaling
A particular feature of pathogenic influence is the striking immunogenetic phenomenon of “stereotyped” BCR structure in CLL, i.e., the occurrence of antigen receptors with a highly restricted repertoire of heavy- and light-chain rearrangement/mutation structure.67 Large-scale studies have identified near-identical BCR sequences (referred to as “stereotypy”) in the variable complementarity determining region among unrelated patients in >20% of cases.68 The BCR similarity between unrelated patients exceeds by far any stochastic reasoning to suggest that these could occur by chance. BCR stereotypy in CLL indicates not only a role for antigen(s) in the pathogenesis and evolution, but it can also be used to group CLL cases into subsets with presumably different biological backgrounds and possibly prognosis,68 again, a relation to be confirmed in clinical trials.
Conversely, antigen-independent BCR stimulation was found in one experimental setting, resulting in cell-autonomous signaling, and it was thought to promote survival.69 Constitutive BCR activation directs many cellular processes, including growth, differentiation, survival, and adhesion or cellular migration, likely attributable to an increased expression of spleen tryosine kinase, LYN, BTK, and PI3K.41,43 Although the exact mechanisms and modulation depend on numerous variables, including the cellular maturation, the (auto)antigen ligation, and the microenvironment, these features may point to future prognostic or predictive markers for the use of agents targeting the BCR signaling pathway (e.g., BTK and PI3K inhibitors).
However, none of these findings regarding BCR stereotypes and autonomous signaling should currently influence patient management, and assessment is needed in well-designed translational projects in the context of clinical trials.
MRD evaluation
The assessment of MRD has become a very important endpoint of prognostic effect in the chemoimmunotherapy setting. MRD positivity was associated with relapse and shorter survival.70 The quantitative assessment of MRD in the CLL8 trial has provided additional insight into the clinical significance of MRD as assessed by 4-color flow cytometry.71 The FCR regimen produced lower median MRD levels compared with FC, resulting in longer PFS, and MRD negativity in itself was of independent prognostic effect on outcome. Therefore, MRD assessment is recommended in clinical trials using standardized protocols of either 4-color flow cytometry or allele-specific oligonucleotide PCR, although evaluation of MRD is currently not recommended for routine clinical practice.1 However, although in the chemo(immuno)therapy era MRD negativity has been associated clearly with improved outcome,71 this concept needs reassessment in the era of signaling kinase inhibitors. It has been demonstrated that persistent lymphocytosis can be associated with improved outcome,53 questioning the reliability of detection of blood disease as a prognostic marker. Therefore, because of uncertainty regarding the timing of response and the effect of combination partners, it appears mandatory to reconfirm surrogate endpoints such as MRD regarding their association with long-term outcome before they can be used to guide treatment decisions in the era of signaling kinase inhibitor-based therapy.
Integration into a comprehensive model, the CLL international prognostic index
The wealth of prognostic markers as established in the chemo(immuno)therapy era and the sometimes conflicting results between different studies make it difficult to generate a unifying picture, in particular in different disease phases.6-22 Importantly, there are efforts under way by an international consortium trying to integrate biological and clinical variables into a universal risk score to determine outcome in all CLL patients (CLL-IPI consortium72 ). This model uses 5 independent prognostic factors for OS (age, clinical stage, 17p− and/or TP53mut, IGHV status, and β2-MG) in a weighted grading to define 4 different CLL subgroups with significantly different OS rates at 5 years (93.2%, 79.4%, 63.6%, and 23.3%, p < 0.001).72 This system clearly holds great promise to refine risk stratification based on staging and single parameters alone. However, it is derived from trials based on chemo(immuno)therapy and may be challenged by the advent of the novel agents that may in themselves require the development of integrated risk scores highlighting the “moving target” nature of the prognostic/predictive factor topic.
More than semantic: “prognostic” versus “predictive” markers
There is a wealth of parameters identifying subgroups with different outcome in CLL, i.e., prognostic markers, but there is a lack of predictive markers. Although the terms “predictive” and “predict” are used frequently to describe the relation between a marker and treatment outcome, this is not reflecting the true value of a predictive marker in a strict sense.73 Predictive markers identify subgroups of patients that benefit from one particular treatment compared with another. Therefore, predictive markers in a true sense can only be identified and confirmed in randomized trials. The value of a predictive marker is the guidance of treatment decisions, and examples include the HER2 status in the light of treatment with trastuzumab in breast cancer or CD20 expression and treatment with rituximab in lymphoma. Such predictive markers in a strict sense are lacking in CLL, with the potential exception of NOTCH1 mutation and PTK2 expression in the context of rituximab addition to FC,22,73 but these data need confirmation in independent cohorts before being applied in routine practice. It may be assumed that 17p−/TP53mut is a predictive marker for the choice of novel agents such as ibrutinib or idelalisib over the use of chemoimmunotherapy, although the superiority has so far not been proven in a randomized comparison. True predictive markers are highly desirable in the era of novel treatment options such as BCR or BCL2 inhibitors in CLL to allow a rational treatment choice, in particular when considering the cost and value of these novel treatment options. Therefore, the identification of truly predictive markers is an ultimate research goal in the era of “precision medicine” and should be a research priority in CLL.
Acknowledgments
This work was supported by the CLL Global Research Foundation (Alliance), the Else Kröner-Fresenius Project (2010_Kolleg24, Project 2012_A146), Virtual Helmholtz Institute Grant VH-VI-404/TP2, the Federal Ministry of Education and Research (CancerEpiSys, PRECISE), and German Research Foundation Collaborative Research Centre 1074 subprojects B1 and B2. I acknowledge the important contributions of the numerous investigators whose work could not be cited because of space restrictions. I thank all patients and their physicians for trial participation and donation of samples, Hartmut Döhner for longstanding support and mentoring, the German Chronic Lymphocytic Leukemia Study Group (Chairman Michael Hallek), and the European Research Initiative on CLL (Chairman Paolo Ghia) for support and collaboration;
Correspondence
Stephan Stilgenbauer, Department of Internal Medicine III, Ulm University, Albert-Einstein-Allee 23, 89081 Ulm, Germany; Phone: 0049+(0)731–500-45521; Fax: 0049+(0)731–500-45925; e-mail: stephan.stilgenbauer@uniklinik-ulm.de.
