
Abstract
B-cell (B-PLL) and T-cell (T-PLL) prolymphocytic leukemias are rare, poor-prognosis lymphoid neoplasms with similar presentation characterized by symptomatic splenomegaly and lymphocytosis. They can be distinguished from each other and from other T- and B-cell leukemias by careful evaluation of morphology, immunophenotyping, and molecular genetics. The clinical behavior is typically aggressive, although a subset of patients may have an indolent phase of variable length. First-line therapy for T-PLL is with intravenous alemtuzumab and for B-PLL is with combination purine analog-based chemo-immunotherapy. New B-cell receptor inhibitors, such as ibrutinib and idelalisib, may have a role in the management of B-PLL, especially for the patients harboring abnormalities of TP53. Allogenic stem cell transplantation should still be considered for eligible patients and may be the only current therapy capable of delivering a cure. In the past few years, many of the molecular mechanisms underlying disease pathogenesis and progression have been revealed and are likely to lead to the development of novel targeted approaches.
Learning Objectives
To be able to use an integrated approach to the laboratory diagnosis of T- and B-cell prolymphocytic leukemia (PLL) and to be able to distinguish these from other lymphoid leukemias
To be able to select appropriate first-line and salvage treatments for PLL
To understand the role of stem cell transplantation in PLL
A landmark study in 1973 identified B-cell (B-PLL) and T-cell (T-PLL) subtypes of prolymphocytic leukemia.1 This rare aggressive subtype of leukemia had been recognized as distinct from chronic lymphocytic leukemia (CLL) by the clinical presentation with splenomegaly and high white blood cell count, the characteristic morphological appearance of the circulating lymphoid cells, and the poor clinical outcome. 2 Despite their similarities, the B-cell and T-cell subtypes can be distinguished from each other by their unique phenotypic, cytogenetic, and molecular features.
The current therapeutic approach relies on the use of monoclonal antibodies with or without purine analog-based chemotherapy, in some cases consolidated with a stem cell transplant (SCT). However, outcome, particularly in T-PLL, remains very poor, with few long-term survivors.
Over the past few years, new molecular techniques have allowed far better understanding of the recurrent mutations involved in the pathogenesis and progression of PLL. In the future, this will help inform a more targeted treatment approach.
Clinical presentation
PLL is rare, accounting for <2% of lymphoid leukemias. In our specialist diagnostic laboratory, where we analyze 8000 samples from patients with hematological malignancies per year, we saw 11 new cases of T-PLL and only 2 of B-PLL in 2014. It affects older adults with a median age in the 60s (61 years in T-PLL and 69 years in B-PLL) and is more commonly seen in males.3 There have been 2 case reports of T-PLL occurring in children, although one of these in a 6 year old had few features to support a diagnosis of classical T-PLL, and it would be helpful to have confirmation that these cases are genetically similar to that seen in adults4,5 Both subtypes of PLL have a similar clinical presentation. Approximately 10% to 15% of patients may be asymptomatic at diagnosis with a persistent “low-grade indolent” phase that may persist for a few years. More typically, patients present with a short history characterized by B symptoms, splenomegaly that is seen in two thirds of patients and is often massive, and marked lymphocytosis (>100 × 109/L in 75% of T-PLL cases). Lymphadenopathy, although present in more than half of patients, is rarely bulky. CNS involvement is rare (<10%) in both subtypes. However, erythematous or nodular skin rashes, peripheral edema, and pleuro-peritoneal effusions may be seen in up to 25% of patients with T-PLL. T-PLL can be seen as a complication of inherited genetic disorders, such as ataxia telangiectasia and Nijmegen breakage syndrome. There are no other clear genetic or environmental predisposing factors.
Laboratory diagnosis
Accurate diagnosis is dependent on full integration of laboratory results, including peripheral blood morphology, immunophenotyping, cytogenetics, and molecular genetics (Figure 1). Given the rarity of these leukemias, it is important to ensure that there is input from an experienced specialist hematologist/hemato-pathologist in the interpretation of these tests.

Integrated diagnosis of PLL showing characteristic morphology, immunophenotyping, cytogenetics, and molecular genetics for T- and B-cell subtypes.
Integrated diagnosis of PLL showing characteristic morphology, immunophenotyping, cytogenetics, and molecular genetics for T- and B-cell subtypes.
Morphology
Prolymphocytes are medium-sized lymphoid cells with basophilic cytoplasms and prominent nucleoli. The B- and T-prolymphocytes may be indistinguishable morphologically. B-prolymphocytes are often of larger size, and T-prolymphocytes may show characteristic cytoplasmic projections or “blebs.” T-PLL also has small cells (in which the nucleolus is less evident) and cerebriform (in which the nucleus is irregular and folded) variants, seen in ∼20% and 5% of cases, respectively. Histology of other tissues, such as bone marrow, lymph nodes, spleen, and skin, may be helpful in supporting the diagnosis, but the key information is usually obtained from extensive analysis of peripheral blood lymphocytes.
Immunophenotyping
Distinguishing between the B- and T-cell subtypes of PLL is achieved readily through immunophenotyping. It can be more challenging to discriminate between PLL and other T- or B-cell lymphoproliferative disorders. In T-PLL, flow cytometry confirms a post-thymic T-cell population (terminal deoxynuclotidyl transferase-negative, CD1A-negative, CD5-positive, CD2-positive, CD7-positive).3 The majority of cases are CD4-positive, with coexpression of CD8 in ∼25% of cases. Only a minority of cases express CD8 alone. Cytoplasmic CD3 is always present, but membrane expression may be weak or negative. Natural killer and cytoplasmic granule markers are consistently negative. Typically, CD7 expression is strong, whereas CD25 may be negative, thus helping to distinguish T-PLL from adult T-cell leukemia and Sezary syndrome. T-PLL patients are also negative for human T-cell leukemia virus type 1.
B-prolymphocytes show a light-chain–restricted clonal population of mature B-cells. The immunophenotypic profile overlaps with other B-cell lymphomas, which characteristically present with splenomegaly and lymphcytosis, such as mantle cell lymphoma (MCL), splenic marginal zone lymphoma (SMZL), and hairy cell leukemia variant (HCL-v), but is usually distinguishable from CLL.6 Although a proportion of CLL cases [CLL with an increase in prolymphocytes (CLL-PL)] may have an increased number of circulating prolymphocytes (<55%), the characteristic immunophenotype of CLL is retained in these cells and is different from that of de novo B-PLL.
Genetics
T-cell receptor (β and/or γ chains) and immunoglobulin gene rearrangements are seen in the respective subtypes, confirming clonality. Conventional karyotyping may still be informative, particularly in T-PLL, in which complex changes are usually present. Recurrent rearrangements in both disorders are well described.
In T-PLL, the characteristic cytogenetic abnormalities are inversion 14 [inv (14)(q11q23)] and t (14;14)(q11q23) seen in 70% of patients, with t (X;14)(q28q11) seen in ∼20%.7 These rearrangements between TCR genes on chromosome 14 and the proto-oncogenes TCL-1 (and TCL-1B) or MTCP-1 (on chromosome X) lead to overexpression of these oncoproteins and, via activation of AKT, result in cellular proliferation and survival.7 Abnormalities of chromosome 8 (trisomy 8, isochromosome 8q) are the next most commonly seen abnormalities. Other recurrent abnormalities seen with conventional techniques include loss of 11q23 (ATM inactivation) together with additional losses (22q, 13q, 6q, 9p, 12p, and 17p) and gains (22q and 6p).8 Combination studies of karyotype, fluorescence in situ hybridization, single nucleotide polymorphism, and gene expression profile data have identified minimally deleted regions, including that of CDKN1B on chromosome 12, which encodes for a protein essential in cell cycle regulation. Over the past year, reports from next-generation sequencing have confirmed the frequent abnormalities in ATM (70%) and chromosome 8 (77%) and identified a number of new abnormalities, particularly in the JAK–STAT pathway.9-11 These include gain-of-function mutations in IL2RG, JAK-1, JAK-3, and STAT-5B, which all lead to activation of STAT-5B. JAK-3 mutations alone have been reported in 30% to 40% of patients. Together, three quarters of patients showed genetic abnormalities within the JAK–STAT pathway. A novel fusion gene, SEPT9–ABL1 has been reported in a single case of T-PLL, which also leads to downstream activation of STAT-5.12 Additional mutations have been identified in EZH2 (epigenetic regulator), FBXW10, and CHEK2 (DNA repair). Deregulation of any of these pathways provides a strong oncogenic stimulus and is likely to make a major contribution to the pathogenesis and/or evolution of T-PLL.
The most consistent genetic changes seen in B-PLL are abnormalities of TP53 (deletion and/or mutation), seen in 50% of cases,13 and abnormalities of MYC in >50% of cases.14,15 These aberrations in MYC are not necessarily associated with aggressive clinical behavior such as is seen in Burkitt lymphoma, and Ki67 expression is often low. Increased MYC rearrangements, for example, t(8;14), and increased MYC-copy number have been identified in a high proportion of the small number of cases studied (in one report, 5 of 6 cases). In some cases, abnormalities of C-MYC and TP53 are seen together. Gene expression profiling has shown a clear distinction between B-PLL, CLL (including CLL-PL), and SMZL but a variable overlap with MCL, particularly in those cases with leukemic presentation.16,17 Conventionally, the demonstration of t (11;14) and expression of cyclin D1 and/or SOX-11 separates MCL from B-PLL, but van der Velden et al17 have suggested that B-PLL represents a subset of MCL, irrespective of the presence or absence of t (11;14). There appears to be a spectrum of B-cell disorders presenting with splenomegaly and lymphocytosis and overlapping morphological, immunophenotypic, and genetic features. Where B-PLL sits within this group of disorders, which include MCL, SMZL, and HCL-v, is less clear, and the issue has not been resolved fully by next-generation sequencing.
Treatment
The rarity of PLL means that there is very little published data regarding treatment. In T-PLL, there have been a few collaborative phase 2 single-arm studies and retrospective analyses. In B-PLL, there have been a few case reports and small series. In neither subtype has there been any randomized clinical trial. There is no treatment licensed specifically for this indication. Recommendations here are thus based on best available data and personal experience.
Watch and wait
In those patients with either subtype, presenting with an indolent prephase, watchful monitoring is a reasonable approach. This situation may persist for a number of years without any clear evidence that early treatment intervention will be beneficial. However, in most cases, progression is inevitable and may occur rapidly, so careful monitoring is appropriate.
First-line therapy
In T-PLL, there is very limited nondurable response to conventional chemotherapy [eg, CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)], with a median overall survival (OS) of only 7 months in historical series.3 The strong expression of CD52 on the surface of T-prolymphocytes led to the initiation of studies using the anti-CD52 monoclonal antibody alemtuzumab in this disease 2 decades ago. Since then, published reports have indicated overall response in three quarters of relapsed refractory patients, rising to >90% in the first-line setting.18 Although progression-free survival (PFS) is significantly improved to >1 year in responders, relapse invariably occurs and there are few long-term survivors. Median overall survival (OS) is <2 years. For this reason, eligible patients should be considered for consolidation therapies, such as autologous or allogenic SCTs. In this series of T-PLL patients treated first line with alemtuzumab, the complete response rate was 81%, two thirds of patients were progression-free at 1 year, and more than one third of patients remained alive at 4 years. A small study examined the use of subcutaneous administration of alemtuzumab and compared this with responses seen with intravenous alemtuzumab also in the first-line setting.19 The overall response rate (ORR) of subcutaneous alemtuzumab was 33% versus 91% with intravenous alemtuzumab (consecutive cohorts, nonrandomized). The intravenous route of administration is thus recommended.
Treatment trials in T-PLL, which have included >10 patients are summarized in Table 1. Other single-agent therapies used in T-PLL include pentostatin,20 nelarabine,21 and bendamustine,22 which all deliver response rates in the order of 30% to 50% but response duration of only a few months. Herbaux et al22 report 15 patients with T-PLL treated with bendamustine, 7 of whom failed front-line therapy with alemtuzumab. The ORR was 53% (20% complete response), with a median PFS of 5 months and OS of 8.7 months, independent of previous exposure to alemtuzumab or other features. A prospective multicenter phase 2 trial investigated the use of a combination chemotherapy regimen [fludarabine, mitoxantrone, and cyclophosphamide (FCR)] induction, followed by alemtuzumab consolidation.23 This trial included 16 treatment-naive patients and 9 previously treated patients. The ORR to FCR was 68%, increasing to 92% after the addition of alemtuzumab. Median OS and PFS were 17.1 and 11.9 months, respectively. Worse outcome (shorter PFS, 10.6 versus 24.8 months) was seen in those with a TCL-1 oncogene mutation. The combination of pentostatin and alemtuzumab has been evaluated in a phase 2 trial showing an ORR of 66%, which was not better than alemtuzumab alone.24 However, it is sometimes helpful to add a purine analog, such as pentostatin or cladribine, to alemtuzumab in those patients who have slow or incomplete responses to antibody monotherapy.
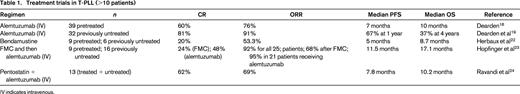
Cases of B-PLL are extremely rare. Most information regarding treatment is derived from case reports or small series (<10 patients). Given the overlap with other mature B-cell leukemias/lymphomas, such as CLL, MCL, and SMZL, the treatment approach has been primarily with regimens developed in these commoner disorders. However, it is recognized that B-PLL does appear to have a poorer survival outcome than CLL. For this reason, suitable patients may be considered for early allogenic SCT, as in T-PLL. The frequent presence of deletions and/or mutations of TP53 explains in part the poor outcome with conventional chemotherapy. However, for the 50% of cases with normal TP53, a conventional chemo-immunotherapy approach with FCR or bendamustine and rituximab is reasonable.25-27 In some reports, an anthracycline has been added, but it is unclear to what extent this improves outcome. Randomized trials in CLL have failed previously to demonstrate superior response rates or PFS with the addition of an anthracycline, and this certainly adds to toxicity. For those patients with nonfunctional TP53, alemtuzumab has historically been the mainstay of treatment and, although no longer licensed in CLL, is available via a patient access scheme.28 More recently, in CLL, the B-cell receptor (BCR) inhibitors (ibrutinib and idelalisib) have been shown to have activity in 17p-deleted CLL, with similar outcomes when compared with those patients with no deletion.29,30 This has led to the licensing of both these agents for first-line therapy in 17p-deleted CLL. In the absence of any prospective clinical trial data for B-PLL, it would seem that BCR inhibitors may be an effective therapeutic option, especially in 17p-deleted cases, and experience with these and other novel agents is likely to emerge over the next few years.
Hematopoietic SCT
In PLL, relapse seems inevitable and remissions are generally of short duration. In addition, B-PLL is often characterized by high-risk genetic abnormalities of TP53. For these reasons, it is appropriate to consider potentially curative treatment with allogenic SCT in first remission for eligible patients. Unfortunately, the age and fitness of patients with PLL often rules out this approach, although reduced-intensity conditioning regimens have widened applicability in recent years. There have been a number of publications of SCT in T-PLL suggesting that this can improve OS and may deliver a cure in a minority of cases31-33 (Table 2). The issues remain the relatively high transplant-related mortality (TRM) and relapse incidence, with less than half of transplanted patients in sustained remissions. Guillaume et al33 reported 27 T-PLL cases from retrospective registry data of allogenic SCT. At the time of allogenic SCT, 14 patients were in complete remission (CR), 10 were in partial remission (PR), and only 3 had refractory disease. With median follow-up of 33 months, 10 patients remain in continuous CR. At 3 years, OS was 36%, PFS was 26%, and TRM was 31%. The relapse incidence was 47%, with a median duration of 11.7 months and all relapses occurring within the first 2 years. These results are better than the European Society for Blood and Bone Marrow Transplantation retrospective data32 but similar to our own study31 , with the key predictor for better outcome being the remission status at the time of transplant. Relapses occur early, usually in the first 2 years, but surprisingly, we have seen recently 2 late relapses in our series of patients at 7 and 12 years after allogenic SCT. Szuszies et al34 reported on 3 T-PLL patients receiving reduced intensity allogenic SCT after induction of a CR with alemtuzumab, who had good engraftment but with subsequent diminishing donor chimerism between days 28 and 290. This was not associated with overt relapse, and full donor chimerism was regained after donor lymphocyte infusion (DLI).

In B-PLL, there are a number of case reports of successful transplants, although inevitably case reports are misleading because they fail to highlight the number of unsuccessful cases.35-37 Kalaycio et al36 report on 11 cases of B-PLL with a median follow-up of 13 months. At 1 year, PFS was 33% and TRM was 28%, which is similar to the outcomes for T-PLL above. They saw no difference between reduced intensity and full myelo-ablative conditioning.
Relapsed or refractory disease
PLL is not curable with chemotherapy and/or antibody treatment, relapse is inevitable, and, in some patients, remission duration is short. In neither subtype has the role of any maintenance therapy been explored. In T-PLL, approximately half of patients will achieve second remissions with alemtuzumab but usually of shorter duration. However, expression of CD52 may be lost at relapse, rendering alemtuzumab ineffective. There is some evidence that epigenetic therapies (eg, histone deacetylase inhibitors), with or without hypomethylating agents such as cladribine, may be able to modify CD52 and other molecules and thus overcome treatment resistance.38 Other salvage therapies include nelarabine21 and bendamustine,22 but these rarely deliver prolonged remissions. Intensive regimens followed immediately by allogenic SCT can be effective but only in a minority. Novel treatments targeting dysregulated pathways, such as JAK–STAT are likely to be the mainstay of future therapeutic trials.
In B-PLL, depending on the remission duration after first-line treatment, relapse can be managed with the same or similar chemo-immunotherapy regimens. Patients with early relapse or that were associated with high-risk genetics (eg, abnormal TP53) may be considered for treatment with novel BCR inhibitors, such as ibrutinib or idelalisib, or other experimental therapies, preferably within a clinical trial setting.
Relapse after allogenic SCT is associated with a dismal outcome. Most commonly, relapses occur within the first 3 years, with a peak incidence in year 1. In some cases, DLI has been effective, and it may be that close monitoring and early intervention may be beneficial to preempt full-blown relapse.
PLL management algorithm
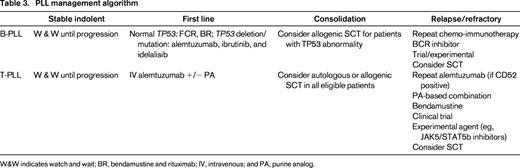
W&W indicates watch and wait; BR, bendamustine and rituximab; IV, intravenous; and PA, purine analog.

Treatment algorithm for T-PLL. IV indicates intravenous; PD/NR, progressive disease/no response; FIC, full intensity conditioned allogeneic SCT; RIC, reduced intensity conditioned allogeneic SCT.
Treatment algorithm for T-PLL. IV indicates intravenous; PD/NR, progressive disease/no response; FIC, full intensity conditioned allogeneic SCT; RIC, reduced intensity conditioned allogeneic SCT.
Summary
PLL comprises two subtypes, T-cell and B-cell, both of which are rare lymphoid malignancies with aggressive clinical course and poor prognosis. Clinical presentation, with splenomegaly and high lymphocyte count, may be similar, but the biology and genetics is quite distinct. Although a subset of patients may have an indolent phase of variable length, progression is inevitable. Treatment is not curative but can deliver high response rates and reasonably durable remissions, measured in years for those achieving CR. For T-PLL, first-line therapy is with intravenous alemtuzumab and for B-PLL is with combination chemo-immunotherapy for patients with normal TP53 and with alemtuzumab or BCR inhibitors for those with deletions or mutations of TP53. Allogenic SCT should be considered for eligible patients. Novel therapies targeting key pathways, JAK–STAT in T-PLL and BCR signaling in B-PLL, are likely to provide new approaches in the future.
Correspondence
Dr Claire Dearden, Department of Haemato-Oncology, The Royal Marsden National Health Service Foundation Trust, Downs Rd, Sutton, Surrey SM2 5PT, UK. Phone: 020 8661 3655; Fax: 020 8642 9634; e-mail: Claire.Dearden@rmh.nhs.uk.
