
Abstract
Multiple myeloma (MM) is a plasma cell malignancy historically defined by the presence of end-organ damage, specifically, hypercalcemia, renal failure, anemia, and bone lesions (CRAB features) that can be attributed to the neoplastic process. In 2014, the International Myeloma Working Group (IMWG) updated the diagnostic criteria for MM to add specific biomarkers that can be used to make the diagnosis of the disease in patients who did not have CRAB features. In addition, the update allows modern imaging methods including computed tomography (CT) and positron emission tomography-CT to diagnose MM bone disease. These changes enable early diagnosis, and allow the initiation of effective therapy to prevent the development of end-organ damage in patients who are at the highest risk. This article reviews these and several other clarifications and revisions that were made to the diagnostic criteria for MM and related disorders. The updated disease definition for MM also automatically resulted in a revision to the diagnostic criteria for the asymptomatic phase of the disease termed smoldering MM (SMM). Thus the current diagnosis and risk-stratification of SMM is also reviewed in this article. Using specific prognostic factors, it is possible to identify a subset of patients with SMM who have a risk of progression to MM of 25% per year (high-risk SMM). An approach to the management of patients with low- and high-risk SMM is discussed.
Learning Objectives
To learn about the recent changes to the diagnostic criteria for MM, and to identify the specific biomarkers that are incorporated into the new disease definition
To recognize high-risk prognostic factors in SMM and to incorporate risk stratification in the management of patients with this disease
Multiple myeloma (MM) is a clonal plasma cell malignancy that evolves from a clinically silent premalignant stage termed monoclonal gammopathy of undetermined significance (MGUS).1,2 Although MM is a classic life-threatening malignancy and MGUS the prototypic premalignant condition with a low risk of malignant conversion, there are patients who saddle the two extremes in whom a clinical conclusion of premalignancy versus malignancy cannot be readily made even by the best laboratory tools.3 These patients are considered to have smoldering multiple myeloma (SMM), an intermediate clinically-defined stage associated with a much higher risk of progression to malignancy (∼10% per year) than MGUS (∼1% per year).4,5 MGUS and SMM are typically asymptomatic, and are differentiated from each other based on the level of the secreted monoclonal (M) protein and/or the extent of clonal plasma cell involvement on bone marrow examination.
The diagnosis of MM traditionally required demonstrable evidence end-organ damage attributable to the neoplastic clone of plasma cells: hypercalcemia, renal failure, anemia, and osteolytic bone lesions, commonly referred to as CRAB features.6 Although this clinicopathologic disease definition ensured that patients with MGUS were not subjected to unnecessary and toxic chemotherapy, it also prevented patients with true early stage malignancy contained within the SMM subgroup from being treated in a timely manner. Although it is clear that some patients with SMM have biologic premalignancy and could therefore be safely observed, there is ongoing concern about the optimal management of patients with SMM who have true malignancy in whom it is only a matter of time before end-organ damage occurred. Unfortunately, there were no reliable ways to distinguish patients with SMM who had premalignancy from those with asymptomatic malignancy.
Revised diagnostic criteria for MM
In 2014, the International Myeloma Working Group (IMWG) revised the diagnostic criteria for MM.7 The update was prompted by the identification of reliable biomarkers that can accurately distinguish patients with SMM who had a high likelihood of true malignancy and were therefore at imminent risk of end-organ damage. It was also supported by improvements in myeloma therapy over the last 10 years, and data suggesting that intervention at the SMM stage in high-risk patients may improve overall survival.8-10 In updating the diagnostic criteria, the IMWG also recognized that continuing to rely on CRAB features as an essential component of the diagnoses not only prevented high-risk patients from receiving therapy to prevent organ damage, but more importantly systematically deprived the administration of novel treatments when the malignancy is most susceptible. In other words, the possibility of cure in MM may not be realized if therapy is always administered only after end-organ damage has occurred at which point the malignancy is extensive, has more cytogenetic abnormalities, and is more likely to be microenvironment independent.3
The revised diagnostic criteria for MM allow the use of specific biomarkers to define the disease in addition to the established CRAB features. They also allow the use of modern imaging tools to diagnose MM bone disease, and clarify several other diagnostic requirements. The update to the disease definition of MM also automatically resulted in revised diagnostic criteria for SMM. In addition to MM and SMM, the IMWG also updated the diagnostic criteria for several other related plasma cell disorders. Table 1 provides the revised IMWG criteria for diagnosis of MM and related plasma cell disorders.7 Table 2 provides a summary of the major changes made in the new criteria.
IMWG diagnostic criteria for MM and related plasma cell disorders
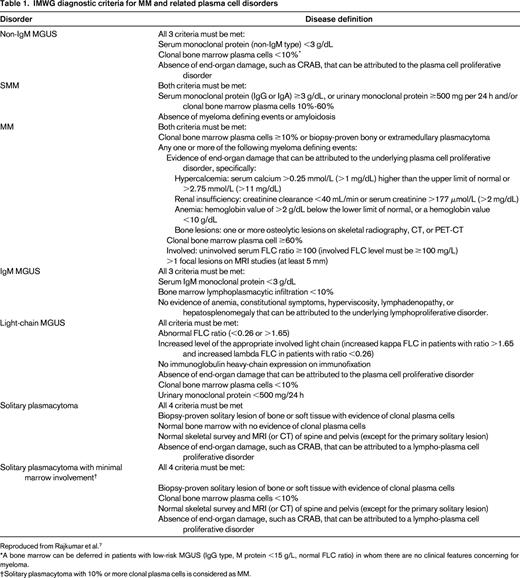
Reproduced from Rajkumar et al.7
*A bone marrow can be deferred in patients with low-risk MGUS (IgG type, M protein <15 g/L, normal FLC ratio) in whom there are no clinical features concerning for myeloma.
†Solitary plasmacytoma with 10% or more clonal plasma cells is considered as MM.
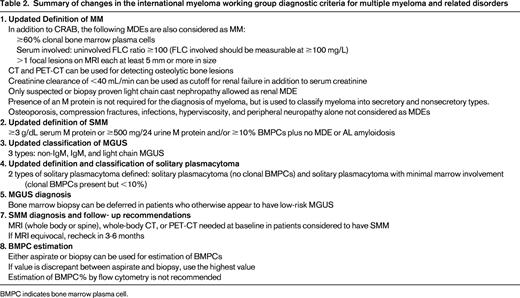
Myeloma defining events
The diagnosis of MM requires the presence of one or more myeloma defining events (MDEs) in addition to evidence of either 10% or more clonal plasma cells on bone marrow examination or a biopsy-proven plasmacytoma.7 MDE includes established CRAB features, as well as biomarkers, that are associated with an ∼80% risk of progression to symptomatic end-organ damage in two or more independent studies. Thus in addition to CRAB features, the following 3 biomarkers that met the prespecified threshold are considered as MDEs: clonal bone marrow plasma cells ≥60%, serum free light chain (FLC) ratio ≥100 provided involved FLC level is ≥100 mg/L, or more than one focal lesion on magnetic resonance imaging (MRI). Patients with these markers were felt to have such a high probability of end-organ damage that delaying therapy until such damage could be documented was not felt to be in the best interest of the patients: in these patients, the disease is not smoldering but is clearly an open flame at this point.
Extreme bone marrow clonal plasmacytosis
Clonal bone marrow plasma cell involvement of ≥60% is extremely unusual in the absence of CRAB features. In the Mayo Clinic cohort of SMM, only 6 of 276 patients (2%) had clonal bone marrow plasma cells ≥60%. In such patients, progression to symptomatic malignancy was rapid; median progression-free survival (PFS) was 7.7 months.8 In another Mayo Clinic cohort of 651 patients with SMM, only 21 (3.2%) had clonal bone marrow plasma cells ≥60%.8 Of these, 95% progressed to MM within 2 years of diagnosis; median time to progression (TTP) was 7 months. A study of 96 patients with SMM from the Greek Myeloma Group also found a markedly high risk of progression in this subgroup of patients, with a median TTP of 15 months.11 Similar results were reported by the University of Pennsylvania; 6 of 121 patients (5%) with SMM had ≥ 60% bone marrow involvement, and all progressed to MM in less than 2 years.12
Marked elevation of serum involved/uninvolved FLC ratio
The serum FLC assay (Freelite, The Binding Site Limited) measures serum kappa and lambda FLCs. The clonal light chain component is referred to as the “involved” FLC (ie, kappa is the involved FLC in monoclonal kappa myeloma, and lambda is the involved FLC in monoclonal lambda myeloma). The ratio of these light chains is almost always perturbed in MM. In SMM, an abnormal FLC ratio is associated with a higher risk of progression to MM.13 Larsen et al found a markedly abnormal involved/uninvolved FLC ratio (≥100) in 90 of 586 (15%) of patients with SMM.9 The risk of progression to MM within the first 2 years with an FLC ratio ≥100 was 72%; the risk of progression to MM or AL amyloidosis in 2 years was 79%. Kastritis et al studied 96 patients with SMM, and found that 7% had an involved/uninvolved FLC ratio ≥100; almost all of these patients progressed within 18 months.11 In a third study, at the University of Pennsylvania, SMM patients with an involved/uninvolved FLC ratio ≥100 had a 64% risk of progression within 2 years.12 To reduce possibility of error, in addition to the FLC ratio ≥100, the IMWG also added a requirement for a minimal involved FLC level of at least 100 mg/L in order to be considered as an MDE.7
MRI with more than one focal lesion
An abnormal MRI imaging study of the whole- body spine/pelvis typically reflects bone marrow changes in SMM, and can be either focal or diffuse. In a study by Hillengass et al, 23 of 149 (15%) patients with SMM had more than one focal lesion on whole-body MRI.14 The median TTP in these patients was 13 months, and the progression rate at 2 years was 70%. These results were confirmed by Kastritis et al found >1 focal lesion on spinal MRI in 9 of 65 patients (14%) with SMM.15 The median TTP was 15 months and 69% progressed to MM within 2 years. The IMWG added a requirement that focal lesions need to be at least 5 mm or more in size, and recommended follow-up examinations in 3-6 months in patients with who had a solitary focal lesion, equivocal findings, or diffuse infiltration.7
Imaging for bone disease
In addition to the skeletal survey, the updated IMWG criteria now allow for the use of computed tomography (CT), low-dose whole-body CT, and positron emission tomography with computerized tomography (PET-CT) to diagnose lytic bone disease in MM. This change will enable early and accurate diagnosis of MM. One or more sites of osteolytic bone destruction of at least 5 mm or more in size is required. Increased uptake on PET-CT is alone not adequate; there must be evidence of actual osteolytic bone destruction on the CT portion of the examination. A biopsy of one of the bone lesions should be considered if there is any doubt about the diagnosis.
Other clarifications
Several other clarifications to the diagnostic criteria for MM were made. These are detailed in Table 2. Of note, the presence of osteoporosis, vertebral compression fractures, or bone densitometric changes in the absence of lytic lesions is not sufficient evidence of myeloma bone disease. In terms of renal disease, only suspected or proven light chain cast nephropathy is considered as an MDE. Other renal disorders associated with M proteins, such as light chain deposition disease, membranoproliferative glomerulonephritis, and AL amyloidosis, are considered unique diseases and not MM. A renal biopsy to clarify the underlying cause of the renal failure is recommended in patients with suspected cast nephropathy, especially if the serum involved FLC levels are <500 mg/L.17 An estimated GFR less than 40 mL/min is preferred to the serum creatinine concentration for purposes of fulfilling the CRAB criteria.
Hyperviscosity, systemic AL amyloidosis, peripheral neuropathy, and recurrent bacterial infections are also not considered as MDEs.
Revised diagnostic criteria for SMM definition
SMM is defined by the presence of a serum M protein of ≥3g/dL and/or 10%-60% clonal bone marrow plasma cells with no evidence of MDE or amyloidosis (Table 1).7 This definition excludes patients previously considered to have SMM with ultra-high-risk of progression (80% within 2 years) who are now classified as MM based on the updated diagnostic criteria. However, this change upstages only a small proportion of patients, and SMM remains a major clinical dilemma with an overall risk of progression of approximately 10% per year for the first 5 years.18 SMM should be distinguished from MGUS, MM, and other related plasma cell disorders using the criteria listed in Table 1.
Molecular classification of SMM
SMM can be subclassified based on underlying cytogenetic abnormalities.20,21 Patients with t(4;14), 1q gain, and/or del(17p) are considered as high-risk SMM (median TTP of ∼24 months). Patients with trisomies are considered intermediate-risk (median TTP ∼34 months); other cytogenetic abnormalities including t(11;14) are considered standard-risk (median TTP, 54 months). SMM patients who have no evidence of cytogenetic abnormalities on fluorescence in situ hybridization (FISH) studies are considered low-risk (median TTP, 101 months).
Dhodapkar et al studied gene expression profiling (GEP) signatures in 331 patients with MGUS and SMM.22 They found that as in MM, a high-risk GEP score (>−0.26) based on a 70-gene signature (GEP70) identifies patients with high-risk SMM. A 4-gene model has also been developed in a study of 105 patients, in which an increased score (≥9.28) was associated with a 2-year-risk of progression of 86%.23
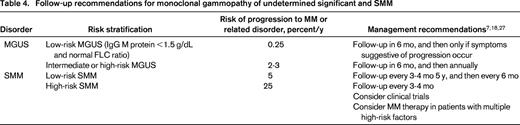
Risk stratification of SMM
The risk of progression of SMM is ∼10% per year for the first 5 years; after 5 years, the risk decreases to 3% per year for the next 5 years, and further decreases to approximately 1% per year thereafter.4 Several studies have identified important biomarkers that can identify patients with SMM who are at high risk of progression (25% per year), and multiple risk stratification models (eg, Spanish and Mayo Clinic models) have been proposed by combining prognostic factors.4,13,14,20-22,24-26 Table 3 provides the criteria for high-risk SMM.18 Although these biomarkers were defined before the revisions to the diagnostic criteria were made, because the proportion of SMM patients affected and upstaged as the result of the new criteria are small, the effect on the estimates for progression would likely be minimal. Identification of high-risk SMM is of particular importance because these patients are at considerable risk of end-organ damage, and are candidates for clinical trials. Moreover, given encouraging results of a Spanish clinical trial in high-risk SMM,10 certain patients with multiple risk factors may even be candidates for therapy after a careful consideration of risks and benefits. In contrast, SMM patients without high-risk factors likely have a risk of progression of 5% per year or less.
Definition of high-risk SMM

Reproduced from Rajkumar et al.18
Note that the term smoldering multiple myeloma excludes patients without end-organ damage who meet revised definition of multiple myeloma, namely clonal bone marrow plasma cells ≥60% or serum FLC ratio ≥100 (plus measurable involved FLC level ≥100 mg/L), or more than one focal lesion on magnetic resonance imaging. The risk factors listed in this table are not meant to be indications for therapy; they are variables associated with a high risk of progression of SMM, and identify patients who need close follow-up and consideration for clinical trials.
*Increase in serum monoclonal protein by ≥25% on 2 successive evaluations within a 6 month period.
Management of SMM
Initial diagnostic evaluation and monitoring.
As discussed earlier, imaging with either an MRI of the spine and pelvis (or ideally whole-body MRI) or whole-body CT/ PET-CT is recommended at baseline to distinguish SMM from MM.7,27,28 Bone-marrow examination with FISH studies and multiparametric flow cytometry are also needed for risk stratification (Table 3). The standard of care for SMM remains observation (Table 4).7,18,27 Patients should be re-evaluated every 3-4 months. In low-risk patients, follow-up can be reduced to once every 6 months after the first 5 years; imaging studies should be done if changes in clinical features or M protein occur. In high-risk patients, follow-up should continue indefinitely, and should include periodic imaging studies to rule out asymptomatic progression. Following the revision to the MM diagnostic criteria SMM patients on observation can be initiated on therapy without waiting for CRAB features to appear if follow-up testing shows the development of other MDE or early detection of MM bone disease based on advanced imaging studies. Patients with a baseline MRI showing diffuse infiltration, solitary focal lesion, or equivocal lesions, need follow-up examinations in 3-6 months to rule out progression.7
Bisphosphonates.
Bisphosphonates have shown promise in the reducing risk of skeletal related events (SREs). In a randomized trial of 177 patients, a reduction in skeletal-related events was noted with pamidronate (once monthly for 12 months) compared with observation, 39% versus 73%, respectively, P = .009.29 However, no improvement in TTP or overall survival was noted. Similarly, in a randomized trial of 163 patients with SMM, a reduction in the rate of SREs was noted with zoledronic acid (monthly for 12 months) versus observation, 56% versus 78%, respectively, P = .041.30 No improvement in TTP was seen. Given the absence of benefit in terms of TTP or overall survival, it is hard to justify monthly bisphosphonates for all patients with SMM given the potential risks of such therapy. However, reduction in SREs is an important endpoint in itself, and more data in this regard would be of value. Until then, I would recommend once-yearly bisphosphonate in low-risk patients, and once every 3-4 months in selected high-risk SMM patients, based on the promising results of 2 randomized trials showing a reduction in SRE with bisphosphonates.18,29,30
Myeloma-specific therapy.
Early studies with melphalan and prednisone (MP) showed no benefit with early therapy.31-33 A phase III trial in 68 patients with SMM found superior TTP with thalidomide plus zoledronic acid versus zoledronic acid alone, median 2.4 years versus 1.2 years, respectively, P = .02.34 However, there were no significant differences in TTP to symptomatic MM, 4.3 versus 3.3 years, respectively, or overall survival, 5-year survival 74% versus 73%, respectively.
More recently the Spanish Myeloma Group reported the results of a randomized trial comparing lenalidomide plus low-dose dexamethasone (Rd) versus observation in 120 patients with high-risk SMM.10 They found significantly longer TTP in patients treated with Rd compared with observation, median TTP not reached versus 21 months, P < .001. More importantly, overall survival was longer with Rd compared with observation, 3-year survival 94% versus 80%, respectively, P = .03. Some limitations of this study include an age difference between the treatment and control groups, the use of a risk stratification model that is not yet widely available, and a study design that is not suitable for regulatory purposes. Nevertheless, this is a landmark study that shows that early therapy may potentially improve overall survival in MM, and was one of the key elements that supported the updated MM diagnostic criteria. Additional studies with lenalidomide are needed, and a randomized trial is currently ongoing in the United States comparing lenalidomide versus observation.
More intensive treatment approaches similar to MM are also being investigated. There is interest in determining if early administration of standard MM therapy to patients with high-risk SMM can prolong overall survival, and may be one of the investigational paths towards a curative approach. Landgren et al treated 12 patients with high-risk SMM with carfilzomib, lenalidomide, and dexamethasone (KRd).35 On interim analysis, 7 of 12 patients (58%) achieved complete response (CR) or stringent CR. Additional patients are being enrolled, and further results are awaited. Trials using triplet induction, stem cell transplantation, and maintenance are being initiated by the Spanish Myeloma Group.
Based on the available data, my approach to the treatment of SMM is summarized in Figure 1. I recommend observation alone for low-risk patients. The risk of progression in these patients is relatively low (∼5% per year) and declines further after the first 5 years of follow-up. These patients are best spared the toxicities of early therapy because the vast majority will be progression-free in 10 years.

Approach to the treatment of patients with suspected smoldering or asymptomatic myeloma.
Approach to the treatment of patients with suspected smoldering or asymptomatic myeloma.
In high-risk patients, the results of the Spanish trial of Rd are provocative.10 The updated MM criteria allowing patients at highest risk to be classified as MM, and the use of advanced imaging tools to diagnose skeletal events early (at baseline and during follow-up) suggest that the risk of serious end-organ damage in this group will be lower than what was observed in the control arm of the Spanish trial. Further, some of the limitations of the trial and the lack of confirmatory studies make it difficult to endorse early therapy for all high-risk SMM patients. We need to determine whether the impact of early therapy apply to SMM patients who are considered high-risk based on factors other than the ones studied in the Spanish trial. We also need to determine whether the effect seen is specific to Rd, or a more general effect of early intervention that can be further amplified by delivering the same treatment as we administer to MM patients. At this point, I prefer that patients with high-risk SMM (Table 3) be offered clinical trials testing early intervention or observed closely. However, given the high risk of progression, selected high-risk SMM patients with multiple risk factors or evidence of biologic progression (rising M protein level) can be considered for MM therapy.18 There are no specific factors to make this determination, and clinical judgment is needed. If therapy is chosen, peripheral blood stem cells should be collected for cryopreservation after approximately 4 cycles of therapy.
Future directions
The updated diagnostic criteria for MM rectified a classic “catch-22” problem that prevented patients with clear-cut malignancy and very high risk of developing end-organ damage from receiving therapy until such damage occurred. The criteria also provide a firm place for advanced imaging in the accurate diagnosis of MM and clarify several diagnostic elements that were either unclear or controversial. Further studies to identify additional reliable markers are needed since the current changes while specific, are not sensitive. We need to identify most patients who are at risk of early progression. Finally, although the addition of new biomarkers to the diagnostic criteria for MM help a small subset of patients, the management of patients with high-risk SMM remains a serious problem but at the same time presents a great opportunity especially with regards to the use of immunomodulatory agents, as well as immune therapies including monoclonal antibodies (eg, daratumumab) and checkpoint inhibitors. Clinical trials in patients with high-risk SMM may provide a path to cure. Thus we need to validate current biomarkers and identify new biomarkers for risk stratification of SMM.
Correspondence
S. Vincent Rajkumar, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; Phone: 507-538-0591; Fax: 507-266-9277; e-mail: rajkumar.vincent@mayo.edu.
