
Abstract
Hemophilia A (HA) and B (HB) are classified as mild (>5%-40%) moderate (1%-5%) and severe (<1%) disease based on plasma factor activity. Severity of bleeding is commensurate with baseline factor levels in general; however, heterogeneity of bleeding in patients is well described. Recurrent bleeding with painful and disabling musculoskeletal complications is the largest source of morbidity for persons with hemophilia (PWH) but treatment advances through the years has led to improved outcomes. In the early 20th century, only whole blood and fresh frozen plasma (FFP) was available to treat bleeding episodes. In 1959, cryoprecipitate was discovered and became an option for treatment of HA in 1965. In the 1970s plasma fractionation led to the first standard half-life (SHL) concentrates. These products ushered in the use prophylactic therapy to prevent bleeding episodes. However, viral contamination slowed the use of prophylaxis until the 1980s when viral attenuation steps increased the safety of plasma concentrates. In the 1990s recombinant concentrates were developed and prophylactic therapy is increasing widely yet not yet universally used. However even with frequent SHL concentrate infusions outcomes are not optimal as PWH spend the majority of time with factor levels below the normal range and are at increased risk for bleeding. In 2014, the first extended half-life (EHL) products were approved for use and have begun to change the landscape of hemophilia care. Challenges of EHL implementation include patient selection, product selection, dose and schedule of infusions, monitoring for safety, efficacy and outcomes, and managing economic aspects of care.
Learning Objectives
To describe the timeline of factor replacement therapy for persons with hemophilia
To describe challenges to extended half-life factor concentrate implementation in the clinical setting
Epidemiology
Hemophilia A (HA) and hemophilia B (HB)are X-linked bleeding disorders and result from decreased or deficient plasma clotting factors VIII (FVIII) and IX (FIX) respectively. Hemophilia A accounts for ∼80% of all cases of hemophilia with an incidence of 1:5000 live male births. Hemophilia B manifests in ∼20% of persons with hemophilia (PWH) and has an incidence of 1:20 000 live male births. In general, the severity of bleeding in HA and HB depends upon the level of circulating clotting factor activity. Patients with levels of >5%-40% are classified as having mild hemophilia, those with levels of 1%-5% as moderate and those with <1% activity as having severe disease. There are an estimated 20 000 PWH in the US and 400 000 worldwide. Although there is often a family history, ∼1/3 of new cases are due to spontaneous mutations. All races and cultures are affected by hemophilia although reporting and access to care vary widely.
Clinical characteristics
Hemophilia A and HB are often clinically indistinguishable and PWH experience common bleeding manifestations. Patients with severe disease suffer from spontaneous bleeding, whereas those with mild and moderate disease more typically bleed with trauma or surgery. In the newborn period, the most common bleeds are intracranial hemorrhage and bleeding from interventions, such as blood draws, immunizations, and circumcision. Older children and adults experience excessive bruising, soft tissue hematomas, mouth bleeds, joint and muscle bleeding, and intracranial hemorrhage. Although bleeding is similar between HA and HB those with HB may have lower bleeding rates, hospitalizations, and decreased factor consumption, and lower hemophilia severity scores.1
Phenotypic heterogeneity
Severity of bleeding is commensurate with the baseline factor levels in general; however, heterogeneity of bleeding patterns in patients is increasing well described.2-5 Interestingly, for individuals with the same baseline clotting factor levels, considerable variability has been described particularly in joint bleeding patterns.3 Age at first joint bleed in those with severe hemophilia varies between the age of 0.2 and 5.8 years with earlier age of bleeding being associated with higher annual clotting factor use and presence of arthropathy (Figure 1).4 What causes this phenotypic variation is unclear but potential modifiers include those regulated by: (1) genetic factors such as causative hemophilia mutation and resultant residual factor activity along with presence of thrombophilia mutations or concomitant bleeding disorders, baseline von Willebrand antigen levels, endogenous thrombin potential, and platelet procoagulant activity; (2) environmental factors, such as patient activity, presence of joint disease, alterations in body mechanics; and (3) treatment related factors, such as access to comprehensive care and clotting factor, age at initiation of prophylaxis, inhibitor status, prescribed treatment dose and regimen, personal and product pharmacokinetics, and adherence to prescribed regimens.6 Identification of the bleeding phenotype with a validated measure, such as the Hemophilia Severity Score, may allow appropriate individualization of care particularly as novel therapeutics become available.2
![Figure 1. Age at first joint bleed for Dutch patients with severe hemophilia born 1968-2002. Median age at first joint bleed was 1.8 years [range, 0.2-5.8s; interquartile range (IQR): 1.1-2.7]. Adapted from van Dijk et al4 with permission.](/view-large/figure/6497099/bep0011505430001.jpeg)
Age at first joint bleed for Dutch patients with severe hemophilia born 1968-2002. Median age at first joint bleed was 1.8 years [range, 0.2-5.8s; interquartile range (IQR): 1.1-2.7]. Adapted from van Dijk et al4 with permission.
Age at first joint bleed for Dutch patients with severe hemophilia born 1968-2002. Median age at first joint bleed was 1.8 years [range, 0.2-5.8s; interquartile range (IQR): 1.1-2.7]. Adapted from van Dijk et al4 with permission.
Historical hemophilia products
Major advances have occurred in treatment products for PWH (Figure 2). Historically transfusion of whole blood was the first available treatment for bleeding episodes. This was largely ineffective because of the low concentration of FVIII or FIX and is no longer recommended. The first reported use of fresh frozen plasma (FFP) in a human for non-hemophilia purposes was 1918. This remained the only option until Judith Graham Pool discovered a method for concentrating plasma into FVIII rich cryoprecipitate in 1959 with subsequent description of large scale production in 1965.7 Cryoprecipitate prepared from single plasma units contains ∼100 IU of FVIII per 8-10 mL and was thus the first viable treatment option for HA. This remains a possible therapy for HA but has been supplanted by virally safer products in developed countries.

Annual number of joint bleeds according to FVIII activity. Black lines are medians; shaded areas are interquartile ranges. Adapted from Den Uijl et al28 with permission.
Annual number of joint bleeds according to FVIII activity. Black lines are medians; shaded areas are interquartile ranges. Adapted from Den Uijl et al28 with permission.
In the 1970s, fractionation methods were developed that produced standard half-life (SHL) plasma-derived lyophilized concentrates of FVIII and FIX. These new products led to the adoption of outpatient treatment for bleeding episodes and the novel approach of prophylactic concentrate infusion in Sweden.8 Primary prophylactic regimens that begin in early childhood are now considered standard of care for those with severe hemophilia in the developed world. However, although thrice weekly prophylaxis for HA and twice weekly infusions for HB are recommended using SHL products, it remains under-utilized in many cases.9
Tragically the majority of persons who received concentrates in the late 1970s and early 1980s became seropositive for HIV and hepatitis C. Because of safety concerns, many patients and providers opted to stay on demand therapy instead of the more factor intense option of prophylactic therapy. In the mid to late 1980s heat treatment, immunoaffinity chromatography, and monoclonal antibody separation steps along with enhanced blood donor testing improved the viral safety profile of concentrates.
Cloning of the FVIII gene in 1982 and the FIX gene in 1984 led to subsequent development of commercially available SHL recombinant factor concentrates in the 1990s.10,11 With increased availability and safety the adoption of primary and secondary prophylactic regimens has continued to increase in developed countries. Multiple iterations of recombinant FVIII (rFVIII) and recombinant FIX (rFIX) products have entered the marketplace since 1993 and each class (FVIII versus IX) have similar pharmacokinetic profiles respectively.12 Safety and efficacy profiles are also similar for these agents.
Extended half-life factor products
Extended half-life (EHL) factor concentrates with different product attributes have been in development for many years and multiple technologies have been used to prolong half-life (T1/2; Table 1).13-18 For FVIII concentrates these include fusion to either albumin or the monomeric Fc fragment of immunoglobin G1 (IgG1). Both extend the half-life of rFVIII by utilizing the neonatal Fc receptor and endogenous IgG recycling pathway. This inhibits lysosomal degradation of IgG and Fc fusion proteins, and recycles them back into the circulation.19 PEGylation is an alternative extension technique. This utilizes a PEG moiety in either a site-directed or a site-specific manner. PEGylation cloaks the molecule in a hydrophilic cloud and delays proteolytic degradation along with decreasing hepatic clearance by binding to low-density lipoprotein receptor-related protein (LRP1). An additional half-life extension technique is to create a FVIII single chain with covalently linked heavy and light chains that have an increased affinity for VWF binding.20
Comparison of extended half-life factor product characteristics
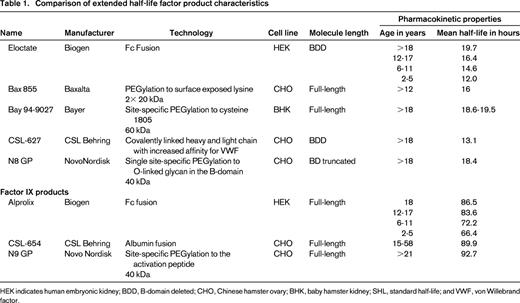
HEK indicates human embryonic kidney; BDD, B-domain deleted; CHO, Chinese hamster ovary; BHK, baby hamster kidney; SHL, standard half-life; and VWF, von Willebrand factor.
The first Food and Drug Administration (FDA) approval of an EHL rFVIII product (Eloctate, Biogen) occurred in 2014. Eloctate is a B domain deleted rFVIII is covalently linked to the Fc portion of IgG1.21 This product has been proven to have a similar IVR modestly increased T½ compared with SHL FVIII products.15,22 Available data suggests that IVR and T1/2 will be similar for additional molecules in development and will likely remain similar until progress can be made to extend T1/2 life of VWF. Alprolix (Biogen) is the first FDA approved EHL rFIX product. Similar to Eloctate, rFIX is fused to the Fc portion of IgG1.23 Other technologies are currently being explored to prolong T1/2 of FIX including PEGylation and albumin fusion.24,25
A basic knowledge of pharmacokinetic (PK) parameters is essential to compare SHL and EHL products and to design and manage factor concentrate regimens. The T1/2 of a drug is the time interval for the concentration of the drug to reach half of its original peak value. A minimum of 2 (peak and trough) but ideally more levels are needed to determine individual PK. For standard factor concentrates, the average T1/2 for SHL FVIII concentrates is 8-12 hours and for SHL FIX concentrates is 12-24 hours. The T1/2 of FVIII is tied to the half-life of von Willebrand factor (VWF) its natural carrier molecule.26 The concentration maximum (Cmax) or peak is the highest drug concentration achieved after a dose. Peak levels may be most relevant during the treatment of bleeding episodes or during invasive procedures. In vivo recovery is a measure of the rise in clotting factor activity achieved after a dose and is represented by the formula:
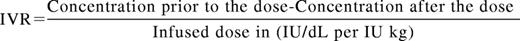
Thus the peak levels achieved for hemophilia products are a result of the number of units of product given and the IVR. The IVR for SHL FVIII concentrates is ∼2 (IU/dL per IU/kg) and for SHL FIX is 1 (IU/dL per IU/kg). Multiple guidelines exist to suggest initial dosing for bleeding events and prophylactic regimens, however, these vary widely from country to country and are dependent upon economic constraints and are rarely evidence based.27 For example a peak of 100% activity is frequently the goal with severe bleeding or injury in resource rich countries but lower peaks are targeted in areas of factor concentrate limitation. The concentration minimum (Cmin) or trough is the lowest drug level achieved prior to the next dose and is likely the most relevant parameter in preventing spontaneous bleeding. Historically, hemophilia providers prescribed prophylactic regimens for those with severe hemophilia aimed to keep trough levels >1%. This was because of the observation that patients with moderate hemophilia (1%-5%) had much lower annualized bleeding rates (ABRs) than those with severe hemophilia. However, the ABR although lower is not negligible in this population (Figure 2).28 The overall importance of peak and trough levels during hemophilia care is not clearly understood however a recent publication suggests that goal trough levels at least in HA may need to be higher than previously thought to eliminate most joint bleeding (Figure 2). Area under the curve (AUC) is the integral of the concentration time curve, which equates to the blood plasma concentration of factor over time. The importance of AUC in bleed control and prevention is increasingly clear as recent publications have shown greater AUC correlates with lower bleeding frequency.15,23
Clearance of factor concentrates refers to the volume of plasma cleared of the drug per over time and is typically higher in younger children and should be considered when designing treatment regimens. The IVR of SHL FVIII is ∼2 IU dL-1 per IU kg-1 and the T1/2 is ∼8-12 hours, but varies greatly amongst patients with ranges of 6-25 hours described The T1/2 of FIX is ∼12-24 hours with an IVR that is roughly 1 IU dL-1 per IU kg-1. With high-dose prophylactic regimens using SHL products persons with moderate and severe hemophilia spend a majority of the time with levels below the normal range and are at increased risk of bleeding and poor outcomes.29 The relatively short half-life of SHL products, the expense and intravenous delivery route are prohibitive to achieving higher trough levels in a majority of patients. Indeed if cost, pharmacokinetics and IV access issues were eliminated it is unclear what the preferred patient factor levels would or should be.
Historical management of hemophilia
Hemophilia management is complex and highly dependent on local resources. Ideal care is multidisciplinary and focused on preventative care. Prior to the advent of effective factor replacement, quality-of-life (QOL) and life expectancy for persons with severe hemophilia was low but now in developed countries approach the normal range. Much advancement in the medical management of hemophilia has occurred in recent decades and the majority has been related to availability of clotting factor replacement (Figure 3). Randomized trials have shown that prophylaxis administration with SHL concentrates improves joint outcomes in children with severe HA compared with episodic on-demand treatment and is considered standard of care.30 Common prophylactic regimens in HA and HB usually involve infusion of SHL FVIII every other day or 3 days per week at 25-40 IU/kg or SHL IX 40-60 IU/kg every 3-4 days. It is increasingly common to use an escalating frequency schedule in young children that entails starting with once weekly infusion, and escalating in frequency as indicated by bleeding rates. Although prophylaxis is superior to on-demand therapy in terms of bleed reduction, frequently patients do not practice prophylaxis as recommended for many reasons.

Evolution timeline of hemophilia A and hemophilia B treatment modalities. Adapted from Rossi's Principles of Transfusion Medicine41 with permission.
Evolution timeline of hemophilia A and hemophilia B treatment modalities. Adapted from Rossi's Principles of Transfusion Medicine41 with permission.
Additional improvements in outcomes have been made because of the availability of comprehensive care and adoption of prophylactic treatment strategies.
Implementation of extended half-life molecules into clinical practice
With the advent of EHL products new challenges face hemophilia providers. The majority of issues surround EHL implementation into clinical practice. These topics surround patient selection, product selection, EHL treatment regimen design, safety and efficacy monitoring, impact on adherence, impact on outcome measures, and management of economic aspects of care.
Patient selection
The first patient-related characteristic to consider is disease severity. Historically patients with severe hemophilia by either residual factor level or bleeding phenotype have been targeted for prophylactic regimens as such individuals have spontaneous bleeds which may be frequent, severe and lead to arthropathy, and require surgical correction. By contrast, those with mild and moderate disease typically have a relatively low ABR, and thus prophylaxis is rarely recommended in this group. The low ABR coupled with cost and limited high level evidence to support prophylaxis in persons with mild and moderate hemophilia mean that most of these patients are managed with on-demand regimens. In addition, many of these less severely affected patients and families are reluctant to perform frequent intravenous infusions with SHL products. It remains to be seen if patients with mild and moderate hemophilia would be amenable to products with less frequent infusion schedules particularly if cost could be justified by improved outcomes.
Another important consideration is the product exposure history of patients. Currently all published data regarding the EHL products have been evaluated in previously treated patients (PTPs) with a minimum of 50 exposure days and no history of inhibitory antibodies. These patients are considered tolerant to factor replacement, and thus far, there have been no published reports of an EHL that has overcome this tolerance in a clinically significant fashion. Animal data has suggested that EHL inhibitor rates may be reduced, however, clinical trials of EHLs in previously untreated human patients (PUPs) are underway or are in developmental stages for the majority of new products, and the information provided on inhibitor development in this population will be crucial in determining safety of these new products. Patients with a history of inhibitory antibodies even those who have subsequently become tolerant have been excluded from clinical research trials of EHLs but are a population well worth investigating and a multicenter trial using EHLs in such patients is in development. Thus far, there are no published reports of immune-tolerance therapy with EHLs but efforts are underway to design these trials in the near future.
Ability to deliver IV factor concentrates is crucial for both SHL and EHL product delivery. Particularly in young children, IV infusions are anxiety provoking for patients and families, and central venous access devices (CVADs) are often used to facilitate infusions. These devices, although effective, are often associated with complications including infection and thrombosis. It is tempting to view the potential of less frequent infusions of EHL products as positive attributes; however, fewer infusions may not lead to better outcomes as it is still unclear which PK parameters are most relevant in the prevention and treatment of bleeding.29 In addition, fewer infusions may impact home infusion proficiency and adherence. Until evidence based guidelines can be developed regarding which PWH will benefit from EHLs, it seems prudent to institute use in patients likely to receive the highest return on investment which are those with severe bleeding phenotypes, no history of inhibitory antibodies and limited IV access.
Product selection
Safety and efficacy are key considerations when selecting a factor concentrate. While there are manufacturing differences, SHL recombinant products are largely similar in pharmacokinetic and safety profiles. Recently several publications have suggested that there may be a difference in terms of safety in inhibitor risk however data has been conflicting.12,31-35 It is likely impossible to retrospectively distinguish small differences in immunogenicity between products as patients are so heterogeneous, and treatment regimens and inhibitor surveillance has not been harmonized. In contrast to the known safety profile of SHL concentrates, the EHL products pose potential new safety issues. Thus far, in published results of EHL products no substantial safety issues have been identified related to Fc coupled, albumin fused, PEGylated or single-chain technology. There is, however, no comparator patient groups in whom long-term safety data is available with these technologies. Continued vigilance for safety signals is warranted when using EHLs in PWH. Ideally, this will be accomplished through continued robust international involvement in EHL clinical trials and post-marketing surveillance.
It is important to recognize that EHL products may not translate to better efficacy in all bleeding scenarios. In particular it is unclear how to treat a bleed that occurs between prophylactic doses of an EHL product although thus far treatment of traumatic bleeds between scheduled prophylaxis doses appears well tolerated.15,16 Surgical experiences with EHL products are beginning to be published, and thus far suggest that surgery can be performed safely with prolonged dosing intervals compared with SHL products.15,36 Further experience with EHL products in the surgical setting is also warranted. Published and presented efficacy outcomes with EHLs are limited to comparison of historical ABRs on SHL products and comparison of ABRs in various EHL demand or prophylactic regimens. Clearly prophylactic regimens are superior to demand regimens; however, it is not clear that EHL prophylactic regimens are superior to SHL prophylactic regimens in terms of ABR. Until sufficient clinical experience is amassed regarding optimal EHL product choice, informed consent conversations between PWH and providers should include discussion of known safety and efficacy data.
Dose and regimen
In general although not identical, current EHL clinical trials follow similar patient selection criteria and trial design. Patients with severe or moderately severe disease (≤2% FIX or <1% FVIII) with no current or historical evidence of inhibitory antibodies have been enrolled on EHL clinical trials. Patients on demand therapy are given a choice between staying on demand therapy and being randomized to secondary prophylaxis. Patients who enter a study on prophylaxis, or those who are randomized to prophylaxis, then choose or are randomized to one or more dose and frequency strata. Dose and frequency is then tailored according to bleeding rates and individual pharmacokinetic data to ensure optimal correction and bleed protection. Per the package inserts of the only FDA approved products, the recommended starting dose of Eloctate for prophylaxis is 50 IU/kg every 4 days and the regimen should be adjusted based on clinical response. The recommended dose of Alprolix is 50 IU/kg once weekly or 100 IU/kg once every 10 days. Insufficient data exists to recommend treatment regimens for any unlicensed EHLs at this point; however, a reasonable approach will be to initiate therapy with FDA approved products according to labeled prescribing information taking into account individual characteristics, such as patient age, activity level, and available vial sizes. Individual PK and close scrutiny of bleeding rates are advised until sufficient population PK data has been amassed to guide other approaches. Allergic reactions are possible with new product exposures so consideration should be given to witnessing at least the first dose of an EHL in the clinical setting with more vigilant observation in patients with known high-risk mutations.
Laboratory monitoring
Monitoring of novel agents includes two major areas of emphasis. The first is the ability to measure EHL factor levels accurately.22 Most laboratories in the US utilize standard APTT based one-stage clotting assays to measure factor levels. This assay result relies upon a contact activator (ellagic acid, silica, and kaolin), calcium, a calibrator (factor-specific or standard), a source of phospholipid, and is run on an analyzer. The final result is determined with either a mechanical or optical clot detection endpoint. All of these variables affect the reproducibility and accuracy of the results of standard and EHL products.37 Factor chromogenic assays detect Xa generation utilizing a chromogenic substrate. There are fewer pretest variables but this assay is not routinely performed in most clinical coagulation laboratories in the US. In terms of EHL FVIII products, in general ellagic acid and some silica-based reagents accurately reflect PEGylated FVIII levels, whereas silica-based reagents are likely to underestimate FVIII levels. FVIII-Fc is generally accurately measured by one-stage assays. Chromogenic assays reliably estimate PEGylated FVIII levels but overestimate FVIII-Fc.38,39 Ellagic acid and silica along with chromogenic assays accurately reflect FIX-Fc activity, whereas kaolin does not. Most activators under or overestimate PEGylated FIX levels, whereas chromogenic assays are generally reliable. Increased communication with the clinical coagulation laboratory with continued guidance by national clinical laboratory organizations is advised when instituting EHL therapy.
Antibodies can develop to any aspect of a protein. Thus far, no high-titer inhibitory antibodies have been reported with EHLs, however, these are novel proteins and routine inhibitor monitoring is prudent. Ideally, inhibitor monitoring would be harmonized within countries but at a minimum within individual HTCs. A reasonable approach to institution of any new factor concentrate is to measure a baseline Bethesda titer followed by repeat measures within a limited number of exposures followed by yearly evaluation.
Outcomes
The major clinical outcomes that have been targeted in PWH include ABR, QOL, and orthopedic outcomes. Additional outcome measures or refined measurement techniques may be needed to determine if EHL products have superior characteristics to SHL products. In particular ABRs and QOL are low in most patients who are compliant with primary prophylaxis with SHL products and it may be challenging to achieve statistically significant improvements in these measures. The impact of EHLs on long-term functional and radiographic joint outcomes will be important to collect as arthropathic processes occur over time, and it will likely take years before the full impact of EHLs on joint health is realized. These observations are not meant to imply that EHLs are unlikely to positively impact outcomes but are instead a reflection of current inability to accurately measure the true impact. Therefore, thoughtful development of outcome tools that reflect the impact of EHLs on PWH are warranted.
Economic
Once an EHL has achieved licensure, reimbursement issues remain and will always be an important consideration in hemophilia care as the large majority of cost of patient care is related to the cost of factor concentrate. Thus far, costs among SHL products have been relatively similar in the US. The pricing strategy for the first to market EHL was to base cost on frequency of use and if prophylactic infusion frequency is reduced 1.5-fold and 2.5-fold, respectively, for FVIII and FIX EHL, then cost will be similar to current cost for SHL. However, if the goal of therapy is to achieve higher trough levels and potentially better outcomes the yearly cost of therapy with EHLs will rise commensurately. In addition, this price structure may limit the feasibility of expanding prophylactic regimens to patients with milder disease phenotypes. Thus, it is reasonable in this initial phase of EHL implementation to limit these products to patients with severe bleeding phenotypes and to utilize PK modeling to control cost.40 Patient input is a critical when it comes to hemophilia care and product choice, and should be incorporated at every decision point.
Conclusion
Hemophilia care is a complex, expensive, and ever-evolving field. With the advent of EHL products, the interactions among comprehensive HTCs, consumers, economic and surveillance organizations, and the pharmaceutical industry will be increasingly important. Whenever possible evidence-based medicine should guide treatment plans. HTCs should consider instituting a plan that incorporates implementation of EHL agents with currently available products along with a research agenda that best meets the needs of the PWH that they serve. Ideally, this plan will include the discussion about newly approved products, pipeline products, and clinical research opportunities that are incorporated into comprehensive visits and community outreach events. An individualized approach to identifying patients with the highest benefit from novel agents should be encouraged. Product dosing and frequency should be grounded in clinical trial outcome data and tailored to individual patient bleeding phenotype and lifestyle. Population PK data and systematic inhibitor monitoring are advised. Ongoing involvement with clinical coagulation laboratory specialists is needed to guide EHL testing. Although great strides have been made over the last few decades, there are multiple hurdles yet to overcome before we can achieve a bleed-free world and harmonization of clinical trial regimens and outcome measures reporting would be welcome by the hemophilia community.
Correspondence
Amy Dunn, Director of Hematology, Director, Hemophilia Treatment Center, The Ohio State University, Nationwide Children's Hospital, 700 Children's Drive, Columbus, OH 43205; Phone: 614-722-3564; Fax: 614-722-3369; e-mail: amy.dunn@nationwidechildrens.org.
