
Abstract
For more than 3 decades, the scientific community has pursued gene correction of hemophilia, with the goal that an individual with congenitally deficient factor VIII or factor IX might synthesize adequate endogenous clotting factor to be relieved of burdensome repeated clotting factor infusions, as well as the emotional weight of continuous hemorrhage risk. Recent reports of successful factor IX gene therapy and partial correction of the bleeding phenotype have raised the bar for success for a robust crop of new clinical gene therapy efforts for both hemophilia A and B. At the same time that gene therapy is gaining momentum, suggesting the possibility of relief from regular intravenous coagulation protein replacement, a number of innovative technologies that enhance hemostatic potential independently of replacement factor administration are demonstrating success in human clinical application. Human clinical trial progress is reviewed regarding a recombinant bispecific IgG antibody to factors IXa and X that mimics factor VIII cofactor activity, as well as monoclonal antibody and short interfering RNA strategies that demonstrate hemostatic efficacy via opposing inhibitors of coagulation. These strategies, associated with prolonged hemostatic potential following subcutaneous (ACE910, ALN-AT3, Concizumab) or single administration (eg, gene therapy) make it possible to imagine a day when recombinant clotting factor administration, rather than being a daily preoccupation, is relegated to an adjunctive role in supporting more novel standard of care therapies.
Learning Objectives
Hematologists and others who are frequently involved in the care of individuals with hemophilia must understand that multiple clinical trials for gene delivery of both factor VIII and factor IX are commencing currently, and should have some broad familiarity with potential benefits and risks of viral vector-based and ex vivo-transduced cell-based approaches to hemophilia gene therapy
Recognize that current limitations of adherence to standard of care intravenous clotting factor concentrate prophylaxis may be addressed in the near future by the adjunctive use or substitution of subcutaneous drugs that increase hemostatic potential with prolonged bioavailability
The hemophilia treatment landscape in 2015
Since the time of the 1965 report of the preparation of cryoprecipitate by Judith Graham Pool and the subsequent availability of FVIII concentrates in the 1970s, intravenous clotting factor concentrates have been the mainstay of therapy for hemophilia. Extraordinary early efforts to elucidate the molecular/genetic underpinnings of hemostasis and the coagulation proteins and their inhibitors enabled the rapid development of recombinant clotting factor concentrates in response to the tragic viral contamination of plasma-derived clotting factors in the 1980s. In part, because routine use of these products in prophylactic replacement therapy can completely change living with congenital severe hemophilia, most new products licensed for hemophilia care in the past 3 decades have involved modifications (primarily additional pathogen removal measures) of recombinant clotting factor concentrates. In the past 1-3 years, new and potentially disruptive investigational products (that are not intravenous clotting factor concentrates) have shown promise in human clinical trials to support deficient hemostasis or to achieve a genetic cure.1-3 This report does not attempt an exhaustive review of all novel therapeutic approaches for bleeding disorders, but instead examines gene therapy approaches and the non-clotting factor, prohemostatic strategies that are in human clinical investigation or that are on a path to translate to human clinical application in the next 1-2 years.
First glimpse of a cure: hemophilia B gene therapy
Despite the investigation of a wide variety of strategies to deliver nucleic acids for hemophilia therapy, human clinical success has only been achieved for hemophilia B, and only via the use of recombinant adeno-associated virus (rAAV) as liver-targeted vector for factor IX gene addition.3 The wild-type virus AAV is a nonpathogenic, replication-defective, single-stranded DNA virus of the genus Parvoviridae, having a genome of only 4680 nt.4 Humans are the natural host for AAV serotype 2 (AAV2); most individuals encounter AAV2 in the environment and raise antibodies that may neutralize subsequent AAV2 infection and may cross-neutralize shared AAV capsid epitopes of other AAV serotypes.5 Targeting the liver using AAV gene therapy has two potential advantages: (1) post-translational processing of the transgenic protein is optimally performed by the liver, the natural organ of factor IX synthesis; and (2) therapeutic factor IX endogenously secreted by the liver after gene therapy has been reported in several animal models to promote clotting factor tolerance.2,6
The pioneering AAV liver-directed, dose escalation gene therapy trial used AAV serotype 2 to deliver a single strand (ss) factor IX cDNA.7 (ClinicalTrials.gov identifier NCT00076557; status terminated.) Although 1 subject at the highest vector dose used in this trial [2 × 1012 vector genomes (vg)/kg] initially produced >11% factor IX, an apparent vector dose-dependent cellular immune response eliminated the AAV-transduced liver cells and factor IX expression. In an effort to avoid the apparent vector dose-dependent hepatocyte elimination that prevented gene persistence, modifications were made to the AAV vector capsid, the AAV genome, and the factor IX cDNA sequence. The AAV2 capsid was changed to AAV8 (natural host rhesus monkeys). The prevalence of AAV8-neutralizing antibodies in humans is lower than AAV2-neutralizing antibodies and AAV8 vectors more selectively transduce liver than AAV2. Following host cell infection, the genome of single-strand AAV (whether wild-type or recombinant vector) must be converted to a double-stranded template for DNA transcription, which is a rate-limiting step in gene delivery.4 By incorporating two complementary copies of the factor IX cDNA into the gene expression cassette carried within the AAV, “self-complementary” AAV (scAAV) vectors demonstrate more efficient gene transfer than ssAAV.8-10 Additional vector efficiency was achieved via codon optimization of the factor IX expression cassette, incorporating nucleotide changes to substitute codons frequently used in the most highly expressed mammalian genes, and making other changes that conserve the factor IX amino acid sequence but improve mRNA translation.11
In the resulting University College London/St Jude Children's Research Hospital (UCL/SJCRH) dose-escalation trial of scAAV8 factor IX reported by Nathwani et al,12,13 all 10 reported subjects express factor IX, which after the first half year following AAV infusion has been stable between 1$-6% activity during follow-up lasting between 2 and 5 years (Table 1). The 6 subjects treated at the high dose (2 × 1012 vg/kg) express a mean 5.1% factor IX activity. Four of the 6 individuals treated at the highest dose did experience asymptomatic mild or moderate liver enzyme elevations, which occurred between weeks 7 and 9 after vector administration. These 4 subjects were treated with 8-12 weeks of prednisolone with rapid resolution of the transaminitis, although the apparent vector dose-dependent CTL response was associated with a relative loss of 50%-70% of factor IX expression in 3 of the 4 subjects.
Multi-year persistence of clotting factor expression following AAV gene therapy in large animals and in humans

IHC indicates immunohistochemistry; and FISH: fluorescence in situ hybridization.
* Adjunctive bortezomib ± dexamethasone administered at the time of AAV vector administration. Follow-up results updated via personal communication from Clinton Lothrop.
† Length of follow-up updated. Oral presentation by A.C. Nathwani at the American Society of Cell and Gene Therapy meeting in New Orleans, LA, May 2015.
Moving beyond the first clinical trial success
Four recent or ongoing liver-directed AAV8.FIX clinical efforts attempt to extend the pursuit of a cure for hemophilia B, that is to say, a therapy that is broadly applicable and delivers long-term expression of clotting factor at levels of >15% activity that would be expected to prevent all musculoskeletal bleeding (Table 2).14 Typically, an abundance of assembled vector capsids that do not carry DNA sequences are generated as a contaminant of recombinant AAV vector manufacture, referred to as empty capsid AAV. Empty capsids comprised >80% of the clinical preparation infused in the reported UCL/SJCRH trial. After incorporating manufacturing steps to decrease empty capsids to <10% of their new clinical preparation, the UCL/SJCRH have treated an additional 2 subjects with 2 × 1012 vg/kg scAAV2/8-LP1-hFIXco (ClinicalTrials.gov Identifier NCT00979238). No hepatic inflammation was observed and these investigators plan a 3-fold escalation in dose for the next subjects in their ongoing trial.
Hemophilia gene and cell therapy programs in late preclinical, IND-enabling, and/or clinical investigations

Also in progress is a phase 1/2 dose-escalation trial of a scAAV8.FIXR338L vector BAX 335 sponsored by Baxalta (ClinicalTrials.gov Identifier NCT01687608).15 The codon-optimized factor IX cDNA incorporated in this vector (previously labeled AskBio009) codes for a single amino acid substitution of leucine for arginine at position 338 of the factor IX catalytic domain. The FIXR338L (Factor IX Padua) has 6-8 times the normal specific activity of wild-type factor IX, and this advantage has been demonstrated by FIXR338L expressed from AAV and lentivirus vectors in mouse and dog hemophilia models.6,16 Baxalta have reported persistent expression of 20%-25% factor IX in at least 1 subject at the second dose (1-1012 vg/kg), in the ongoing trial.17,18 Following dose escalation to the third dose (3 × 1012 vg/kg), fully corrective factor IX expression to >50% activity was observed, followed by liver transaminase elevation associated with immune activation. Factor IX expression was lost despite initiation of corticosteroids in response to the liver inflammation. In light of the unprecedented factor IX expression that was achieved, a prophylactic approach to suppress immune activation is being investigated for the remaining enrollment of up to 16 total subjects.
It is clear from preclinical modeling and human trials that environmental exposure to wild-type AAV results in highly prevalent AAV-neutralizing antibodies and that the presence of even quite low titers (≤1:10 or lower) of pre-existing AAV serotype-specific neutralizing antibody can abrogate any gene expression from systemically-delivered AAV vectors. The BAX 335 (scAAV8R338LFIX vector) sponsors have reported that ∼1/3 of potential subjects screened have been excluded due to pre-existing AAV8 NAbs, despite the fact that macaques rather than humans are the natural host of this serotype. The LP1-hFIXco factor IX expression cassette that was previously delivered in the UCL/SJCRH vector is being packaged in a scAAV serotype 5 vector for liver-targeted expression in a trial sponsored by UniQure. The reported seroprevalence of AAV5-neutralizing antibodies differs widely in various epidemiologic studies (from ∼3% to 36%; Table 3).5,19-22 Of interest is that Nathwani et al have previously compared the LP1-hFIXco sequence packaged in AAV5 versus AAV8 in non-human primates and have shown that although the degree of liver transduction is similar, significant differences exist in particular in the amount of gene delivery by AAV5 to off-target sites such as spleen, kidney and testis.10 Screening and enrollment in the UniQure trial (ClinicalTrials.gov Identifier NCT02396342) began in 2015 for treatment of 5 subjects each at doses of 5.0 × 1012 vg/kg and 2.0 × 1013 vg/kg AAV5-hFIX.23
Prevalence of neutralizing antibodies to AAV in different populations: percentage of subjects with serum inhibiting transduction by naturally occurring serotypes of AAV
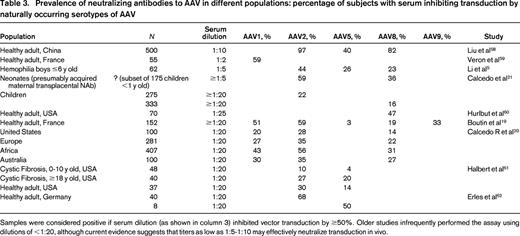
Samples were considered positive if serum dilution (as shown in column 3) inhibited vector transduction by ≥50%. Older studies infrequently performed the assay using dilutions of <1:20, although current evidence suggests that titers as low as 1:5-1:10 may effectively neutralize transduction in vivo.
A single-strand AAV8.FIX clinical trial sponsored by the Children's Hospital of Philadelphia (CHOP) has also treated three subjects (Original ClinicalTrials.gov identifier NCT01620801). Persistent factor IX expression has been observed in 1 of the 3 subjects, whereas 2 subjects lost factor IX expression coincident with evidence of a T-cell response directed against the virus capsid. Spark Therapeutics has announced plans to take forward the CHOP technology in a future clinical trial using immunosuppression, a FIXR338L transgene, and a bioengineered AAV capsid. Together, these recent clinical trial efforts suggest that the understanding of how to consistently predict and to avoid immune elimination of transduced liver remains a major challenge to AAV gene therapy for hemophilia. Ongoing efforts to minimize vector exposure are a logical extension of the approaches used in the UCL/SJCRH and BAX 335 vectors, as exemplified by a recently reported computational strategy to derive hepatocyte-specific transcriptional cis-regulatory modules (CRMs) which achieved 11-15-fold enhancement of liver-specific promoter/enhancer driven factor IX expression in hemophilic mice.24
Sangamo and Shire in collaboration will seek new drug designation in 2015 for a gene editing therapy using AAV to deliver zinc finger nuclease (ZFN) to target genomic insertion and gene addition of the wild-type factor IX cDNA.25 The planned clinical approach will target ZFN cleavage to a site downstream of the constitutively active albumin promoter, to co-opt the synthetic drive of this strong promoter at a locus that they propose is a “safe harbor” for homologous recombination of the F9 gene. Using this approach, the company has reported >3% of normal human FIX level achieved in non-human primates, and a ZFN-targeted factor VIII gene editing program is being developed for human translation in parallel with the factor IX program.26
Additional hemophilia B preclinical development programs have been announced, however, for many of these the timeline for translation to the clinic is difficult to ascertain from publicly available information (Table 2).
Lentivirus (LV) vectors for expression of factor IX have now progressed to cures in hemophilic mice and hemophilic dogs.16 An early concern with the use of lentivirus for the delivery of factor IX or factor VIII was that the broad infectivity of LV permitted transduction of antigen presenting cells and loss of immunologic tolerance for the clotting factor protein (ie, inhibitor development). Following liver- restricted expression using both liver-specific transcriptional regulatory elements and hematopoietic microRNA target sequences to oppose gene expression in APCs, inhibitor development is avoided and LV expression of factor IX is capable of reversing pre-existing factor IX inhibitor antibodies, via the induction of CD4+ CD25+ FoxP3+ regulatory T cells.27 The timeline for systemically delivered liver-directed LV factor IX vectors to translate to clinical application is unclear, although the application of this strategy to both hemophilia B and hemophilia A is the translational goal of a partnership between the San Raffaele Telethon Institute for Gene Therapy (TIGET) and Biogen.
Factor VIII gene therapy returns to the clinic
Initial strategies employed in human clinical trials for hemophilia A gene therapy included factor VIII gene delivery using systemically administered viral vectors (retrovirus or “gutless” adenovirus) and omental implantation of ex vivo transduced fibroblasts autologous fibroblasts. No sustained factor VIIII responses were seen in any trial and no subjects have been enrolled in hemophilia A gene therapy trials in the past 12 years. Nevertheless, the successful clinical application of AAV for the correction of factor IX has encouraged new efforts to harness AAV for F8 gene delivery. The B-domain–deleted (BDD) factor VIII cDNA is 4.4 kilobases (kb), meaning that a BDD FVIII expression cassette having even minimal transcriptional regulatory elements (eg, promoter, polyadenylation site, etc) along with the two 145 nt AAV-inverted terminal repeats (required for packaging the therapeutic gene sequences) exceeds the normal packaging constraints of wild-type AAV (genome 4.7 kb). Incremental increases in the size of the therapeutic sequences beyond ∼ 5.2 kb have been associated with incrementally diminished yields of recombinant AAV vector, with the packaging of partial (truncated) genomes, and with decreased efficiency of transgene expression.28
The UCL/SJCRH investigative team reported in 2013 the use of AAV8 to express codon-optimized BDD FVIII transgenes driven by small liver-specific promoters. [Unlike factor IX, the natural site of production of factor VIII is not hepatocytes, but rather the liver sinusoidal endothelial cells (LSECs). Nevertheless, factor VIII expression from hepatocytes following gene transfer appears to result in factor VIII protein with relatively normal specific activity.] A short sequence derived from juxtaposed B domain elements, resulting in the expression of BDD FVIII with a 17 amino acid peptide encoding 6 glycosylation triplets (v3 sequence), has been incorporated with the goal of improving secretion of factor VIII. In vivo delivery of this rAAV8-HLP-codop-hFVIII-v3 vector achieved physiologic and supraphysiologic factor VIII levels in factor VIII knockout mice, resulting in protection from bleeding.11,29 When used at the vector dose of 2 × 1012 vg/kg (equivalent to the highest dose used in the UCL/SJCRH hemophilia B trial) stable factor VIII expression of 15% was observed in a non-human primate. The use of this FVIII expression cassette has been licensed for clinical use by BioMarin. It is currently anticipated that a human clinical trial of rAAV8-HLP-codop-hFVIII-v3 will commence in early 2016 sponsored by the academic partnership of UCL with SJCRH, and will enroll simultaneously with a BioMarin trial, to commence in 2015, of the same HLP-codop-hFVIII-v3 therapeutic sequences packaged within an undisclosed AAV capsid variant (BMN 270). Baxalta also plans to commence an AAV8 factor VIII gene therapy trial in 2016; details of the candidate FVIII gene expression cassette have not been disclosed. Additional groups have stated that they will pursue investigational drug approval in 2016 and hemophilia A trials using AAV-based approaches including Spark, Dimension/Bayer (likely using AAV serotype 9 or 10), Uniqure (AAV5), and Sangamo/Shire (ZFN gene editing; Table 2).
Lentivirus gene transfer for hemophilia A
Lentivirus vectors efficiently transduce dividing and non-dividing cells and additionally appear to have lower genotoxicity than γ-retroviral vectors, in part because they are less likely to insert near promoters of active genes.2 For direct transduction of the liver, LV in general have led to less robust expression than several serotypes of AAV. In addition, while the incorporation of liver-specific promoters and of microRNA sequences to eliminate transgene expression in hematopoietic antigen presenting cells proved adequate to circumvent inhibitor development of factor IX transgene product, specific pseudotyping of the vector envelope and transient macrophage depletion were required to achieve meaningful factor VIII expression in mice. Additionally, ongoing concern about the oncogenic potential of integrating vectors has led several groups to explore the use of integration-defective lentivirus (IDLV) vectors.30 The liver can express clotting factor from episomally maintained LV-delivered transgenes, but with somewhat diminished expression when compared to integrated LV-delivered transgene. The Telethon Institute for Gene Therapy (TIGET)/Biogen partnership report that their program will use LV to target hepatocytes, however, the timeline for clinical translation is not clear.
Lentivirus ex vivo gene transfer in cell-based therapy for hemophilia correction
Progress continues in the use of integrating lentiviruses to deliver clotting factor genes to pluripotential and multipotential stem cells.31,32 Recent proof-of-concept studies suggest that underlying gene defects, eg, the common F8 intron 22 inversion mutation, can be corrected in iPSC ex vivo using transcription activator-like effector nuclease (TALEN) or other nucleases, and that the nuclease-mediated genome editing does not negatively affect iPSC pluripotency.33,34 Many safety and efficacy hurdles must be met before iPSC technology is ready for clinical application. In contrast to iPSCs, extensive preclinical work supports the therapeutic potential of autologously harvested hematopoietic stem cells (HSCs). HSCs transduced with lentivirus-factor VIII vector and expanded ex vivo maintain multilineage potential after being re-infused into the donor, with the potential to engraft as a self-renewing population of factor VIII-expressing cells with direct access to the bloodstream. Investigators from the gene therapy program at Emory University, Aflac Cancer and Blood Disorders Center are conducting final studies to support submission of an Investigational New Drug application to the FDA in late 2015, in support of a clinical trial for hemophilia A. The trial is designed to use lentivirus to transduce CD34+ cells harvested from mobilized peripheral blood mononuclear cells with a chimeric human/porcine factor VIII gene. The chimeric HP transgene (>90% human sequence) is engineered for high expression via incorporation of selected sequences of the porcine factor VIII gene that convey reduced engagement of the unfolded protein response pathway. The FVIII-transduced HSCs carry no survival advantage within the host bone marrow, and so transduced HSCs will be transplanted into patients after a non-myeloablative conditioning regimen to generate a niche for engraftment and expansion.
Another HSC-based strategy that is being developed by several laboratories focuses lentivirus-delivered factor VIII and factor IX expression in a more lineage-specific fashion by targeting transgene expression to the megakaryocyte/platelet lineage.32,35-38 Activated platelets mediate the primary response to hemorrhage, respond to initiating events in secondary hemostasis and provide the phospholipid surface for the formation of the factor tenase complex, supporting amplification of thrombin generation. Granulocyte colony stimulating factor (GCSF)-mobilized autologous peripheral blood or bone marrow-derived CD34+ stem cells transduced with lentivirus encoding human B-domainless factor VIII under the control of platelet specific GPIIb promoter (the promoter for ITGA2B gene) express FVIII ectopically during megakaryopoiesis, which is stored in platelet α-granules along with von Willebrand factor (VWF) and released at sites of injury. These LV-transduced CD34+ stem cells then can be transplanted into the hemophilic animals following some bone marrow conditioning to create a niche for engraftment. Although no factor VIII activity is measurable in the circulation, the transgenic clotting factor appears to support platelet surface thrombin generation at the site of injury. The approach has been used to correct the bleeding phenotype in hemophilia A mice and hemophilia A dogs, with >2.5 years of follow-up in the large animal model. One consideration is that the busulfan-conditioning regiment that has been used involves nonmyeloablative doses, but does involve risk of bleeding, so that the dogs received supportive care with FVIII replacement and aminocaproic acid until transgenic FVIII expression could be confirmed. Intriguingly, the platelet factor VIII/VWF expression has been shown to provide hemostasis in the presence of circulating factor VIII inhibitors. A similar approach using LV delivery of GPIIb promoter/factor IX transgene has been demonstrated to result in factor IX storage in platelet α-granules and hemostatic protection of transplanted hemophilia B mice (although hemostasis in the presence of factor IX inhibitors was not seen). The Blood Center of Milwaukee investigators plan a phase I clinical trial of HSC therapy targeting coagulation factor VIII expression within platelets. This team with National Institutes of Health (NIH) support has produced GMP qualified vector and are completing late preclinical (IND-enabling) studies. Recently, a similar LV strategy that uses a platelet integrin 1bα promoter to drive factor VIII expression and alpha granule packaging in transduced HSC was reported to correct the bleeding phenotype of hemophilia A mice following direct intraosseous cell delivery without preconditioning.39 It is not yet established whether this less invasive approach can translate to a larger host.
Replacing factor VIII's function without replacing factor VIII: ACE910
Factor VIIIa cofactor activity is estimated to increase the Vmax of the proteolytic cleavage of factor X to factor Xa by calcium- and phospholipid (PL)-associated factor IXa by 20 000-fold. This co-factor activity is achieved by the coordinated binding of the light chain and A2 subunit of FVIIIa to the light and heavy chains of FIXa, respectively, whereas the FVIIIa A1 subunit binds the FX heavy chain in the PL-bound complex. The distance between the FVIIIa binding sites for FIXa and FX is similar to the distance between 2 antigen-binding sites of human IgG. A recombinant humanized asymmetric bispecific IgG antibody to factors IXa and X was generated and characterized with regard to factor VIII mimetic cofactor activity, supporting plasma thrombin generation. Among the potential advantages of an IgG having VIII mimetic cofactor activity are the low immunogenicity, the typical plasma half-life of 2-3 weeks and the high subcutaneous bioavailability of humanized antibodies. The bispecific antibody ACE910, when used as a weekly subcutaneous injection, has demonstrated in a long-term primate model of acquired hemophilia A to normalize the monkey aPTT and to protect against the development of hemophilic joint hemorrhage compared with outcomes in vehicle-treated acquired hemophilia monkeys. In a first-in-human trial of 18 subjects with congenital hemophilia A, once weekly subcutaneous ACE910 for subjects with congenital hemophilia was well tolerated at doses of 0.3 mg/kg, 1 mg/kg, or 3 mg/kg.40 It is notable that two-thirds of the subjects had factor VIII inhibitors (range, 3-111 BU/ml) at enrollment. Nevertheless, individuals without and with inhibitors exhibited a similar reduction in annualized bleeding rate (ABR). With a median of 9.5 months follow-up (n = 16) subjects given the 1 mg/kg or the 3 mg/kg doses experienced a 90%-100% reduction in the ABR without the need for concomitant intravenous prophylactic clotting factor (eg, IV FVIII or bypassing agent therapy).
Opposing inhibitors of coagulation: inhibiting tissue factor pathway inhibitor (TFPI) and antithrombin (AT III)
The initiation of coagulation consisting of TF/FVIIa-mediated FX activation is regulated by immediate negative feedback via TFPI. In the setting of hemophilia, the potential for amplification of coagulation is deficient, and the action of TFPI to provide tight negative feedback of the initiation of coagulation may contribute to the hemophilic bleeding phenotype.41 Inhibition of TFPI by high-affinity peptides targeting specific TFPI domains, by non-anticoagulant sulfated polysaccharides (eg, fucoidan) and by aptamers have all demonstrated the ability to improve coagulation in hemophilia plasmas. A humanized anti-TFPI monoclonal antibody manufactured by recombinant expression (mAb 2021, concizumab) binds TFPI with high affinity at a predicted binding site that overlaps and interferes with the predicted site of complex formation between TFPI and FXa, thereby interfering with TFPI inhibition of factor Xa. Hemostatic efficacy lasting at least 1 week was demonstrated in an induced hemophilia rabbit model and supported the initiation of a recently reported human clinical safety and pharmacokinetics trial performed in healthy volunteers and patients with hemophilia.42 A wide-dose range of administration intravenous (0.5-9000 μg/kg) and subcutaneous (50-3000 μg/kg) was observed with no product-related serious adverse events. Dose-dependent procoagulant effects were evidenced by increases of D-dimers and prothrombin fragment 1 + 2 without evidence of DIC. TFPI levels in plasma fell in a concentration-dependent manner at IV doses ≥250 μg/kg and at subcutaneous doses ≥1000 μg/kg. Additionally, when added ex vivo to plasma treated with factor VIII NAb, the presence of concizumab resulted in increased thrombin generation. The results justify efficacy studies of concizumab to potentially serve as a long half-life, subcutaneously-administered hemostatic agent. In addition, a first-in-human program is reportedly in development by Bayer HealthCare for a human monoclonal anti-TFPI antibody (currently designated BAY1093884), which is expected to support hemostasis in a manner similar to concizumab.
Aptamer technology takes advantage of the property that stretches of nucleic acids fold to take on structure and shape and may thereby act as protein ligands (independent of any biologic role in transcription or translation). Pools of random-sequence oligonucleotides can be selectively panned against target proteins or sequences using SELEX (Systematic Evolution of Ligands by EXponential enrichment) methodology to generate RNA aptamers with target affinity and specificity comparable with antibodies. Inherently, RNA aptamers have a short circulating half-life (minutes) and have been conjugated to polyethylene glycol (PEG) for clinical application. With the advancement of a FIXa aptamer into clinical testing in thousands of subjects, an incidence of PEG-associated allergic reactions of ∼0.6% has been associated with the intravenous route of administration. Although the PEG-associated allergy appears lower (and the efficacy of the aptamer longer) using the subcutaneous route of administration, cautious observation seems to be appropriate as PEG-conjugated factor VIII products for intravenous use move from relatively small pre-licensure studies into wider use post-licensure. It should also be remembered that binding a target protein, whether by a high-affinity antibody or a high-affinity aptamer, does not necessarily lead to protein clearance or inactivation, as previously demonstrated by the isolation of anti-factor IX antibodies that enhance factor IXa protease activity (also subsequently refined in the development of ACE910).43 High-affinity aptamer binding is not achieved by rational targeted design but by repetitive rounds of selective evolution. Recently a PEG-conjugated TFPI-specific aptamer BAX 499 (ARC19499) advanced to human clinical trial, based upon animal testing in which the aptamer effectively inhibited TFPI-negative feedback of the initiation of coagulation. Unfortunately, the human phase 1 trial showed an unexpected increase in plasma TFPI levels and reduction in thrombin generation. The apparent cause was aptamer binding to the Kunitz 3 C-terminal domain of TFPI and inhibiting TFPI clearance. Cueing off this experience, 2 peptides targeted to Kunitz 1 and Kunitz 1-2 domains (but avoiding Kunitz 3 interaction) have been linked in a single fusion peptide currently being developed for therapeutic TFPI inhibition.44,45
RNA interference (RNAi) is an ancient cellular machinery that defends against viruses and transposons. For therapeutic application, short double-stranded RNA (dsRNA) can be designed to effectively target and post-transcriptionally interfere with endogenously expressed RNA sequences (short interfering RNA, siRNA), effectively knocking down translation of the targeted protein. The investigational therapeutic drug ALN-AT3 is a siRNA that targets coagulation inhibitor antithrombin (AT) mRNA; the siRNA is conjugated to N-acetylgalactosamine (GalNAc)-cluster molecules that facilitate high-affinity asialoglycoprotein receptor uptake in hepatocytes. In non-human primates, the effect of subcutaneously delivered ALN-AT3 to reduce plasma AT was demonstrated to be both titratable (from 100% of normal at baseline to levels as low as 0% in a dose-dependent fashion) and fully reversible.46 Non-human primates were then treated for 6 weeks with ALN-AT3 0.5 mg/kg weekly and subsequently infused with anti-factor VIII antibody to induce a hemophilia A inhibitor phenotype; the passive transfer of factor VIII inhibitory antibody led to a >60% inhibition of thrombin generation in this monkey model. In contrast, induced hemophilia monkeys treated with ALN-AT3 demonstrated an 80% reduction in circulating AT, associated with normalization of peak thrombin generation. Following 3 weekly subcutaneous injections, healthy human volunteers experienced durable AT knockdown lasting >60 days. A multidose human trial in hemophilia (without inhibitors) nears completion. At higher doses that accomplish >50% knockdown of AT, increases in mean peak thrombin generation of 112 ± 38% and durable improvements in clot formation measured by ROTEM have been seen in the hemophilia trial; the extent to which this degree of correction impacts bleeding rates has yet to be reported. Of additional interest is the report that the thrombin generation profile of human plasmas deficient in factors V, VII, or XI are improved by AT depletion, suggesting potential application of AT RNAi for these more rare bleeding disorders.47
Summary
The immediate future will see the development in parallel of multiple new investigational agents designed to provide a genetic cure for bleeding disorders or to support hemostasis in a manner that may augment or even circumvent the need for repeated clotting factor concentrate infusions. Enthusiasm is tempered somewhat at this stage because only a minority of individuals with hemophilia can qualify at this point for trials using FIX or FVIII adeno-associated gene therapy vectors; excluded are individuals with history of inhibitors (up to 35% of severe HA), with mild/moderate disease, with active hepatitis (up to 50% of men >30 years), and with pre-existing AAV8 NAb (up to 25%-35%).3,48-51 Some of these restrictions will be temporary or overcome. Not explored clinically as yet is the observation that expression of clotting factor from the liver after gene therapy appears to have a tolerizing effect. Each of the innovative subcutaneous drugs in development also may have extended utility for inhibitor patients. Most optimistically, the underserved population of individuals with inhibitors may ultimately be best served by the new technologies.
Acknowledgments
This work was supported by NIH National Heart, Lung, and Blood Institute 1RC3HL103396 and NIH National Heart, Lung, and Blood Institute P01-HL112761. Tong Gui gave invaluable support with paper preparation.
Correspondence
Paul E. Monahan, Gene Therapy Center, 7119 Thurston-Bowles, CB 7352, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; Phone: 919-962-3285; Fax: 919-966-0907; e-mail: paul_monahan@med.unc.edu.
