
Abstract
Currently available evidence supports the contention that elevated levels of factor XI (fXI) are associated with a greater risk of venous thromboembolism and ischemic stroke, but, less convincingly, with myocardial infarction. Conversely, reduced plasma levels of fXI seem to offer some protection from venous thromboembolism and stroke, but not myocardial infarction. Factor XI-deficient patients are at risk for certain types of bleeding, particularly posttraumatic hemorrhage on mucosal surfaces where there is a high endogenous fibrinolytic activity. In contrast, the situation with fXII in human thrombosis remains enigmatic. Deficiency of fXII is clearly not associated with any bleeding risk, but neither does it seem to be protective against thrombosis. The longstanding debate as to whether partial fXII deficiency represents a risk factor for thrombosis remains unresolved, with seemingly conflicting results depending on study design, type of assay used, and analyte evaluated. The possibility that elevated fXII levels represent a risk factor for thrombosis is not borne out in the literature.
Learning Objective
To understand that high levels of factor XI are associated with thrombosis, but that the relationship between factor XII and thrombosis is not established
Introduction
1953 was a landmark year in the history of the intrinsic pathway of blood coagulation. Rosenthal et al described a factor that, when deficient, was associated with a familial bleeding disorder. He noted that both sexes could be affected and that the abnormal clotting times were corrected upon mixing with whole blood from patients with either type of hemophilia (A or B).1 He therefore named this disorder “hemophilia C” and the missing clotting factor “plasma thromboplastin antecedent,” now known as factor XI (fXI). In the same year, Brinkhous et al invented the partial thromboplastin time (PTT) test,2 which was later modified by Rapaport to include a kaolin activation step, which became the basis of today's activated PTT (aPTT) test.3 Two years later, Ratnoff and Colopy described 3 individuals, 2 sisters and a 37-year-old man named John Hageman, who also manifest prolonged whole blood clotting times.4 Unlike Rosenthal's cases, these individuals had no abnormal bleeding history and the discovery of their defect was entirely serendipitous during presurgical screening. This defect was also shown to be hereditary and became known as the Hageman factor, which was later called factor XII (fXII). Further milestones, including the discovery of the last remaining “major” procoagulant, namely fX (“Stuart Prower factor”) in 1956,5 laid the groundwork for the waterfall or cascade models of blood coagulation proposed by 2 groups on either side of the Atlantic.6,7 In this paradigm, the causes of prolongation of the screening tests of coagulation, namely the PT and aPTT, could be fully explained. The autoactivation of fXII after contact with negatively charged surfaces was proposed to be the initiating event that preceded activation of fXI, thereby providing an alternative pathway for the triggering of coagulation via the so-called intrinsic pathway. However, it was recognized quite early that deficiencies of fXII, high-molecular-weight kininogen or pre-kallikrein were not associated with a bleeding phenotype despite their ability to prolong the aPTT, sometimes very strikingly. The fact that even severe fXII deficiency is not associated with bleeding, whereas fXI deficiency of a more moderate degree is, provides compelling evidence for the existence of an alternative mechanism by which fXI is activated during physiological hemostasis. The discovery that thrombin fulfills this role was a milestone observation and led to a revised coagulation schematic in which hemostasis is initiated by tissue factor/fVIIa, with thrombin providing feedback activation of fXI.8 Such a revised model adequately addresses the dichotomous bleeding phenotypes in fXI and fXII deficiencies.
Additional roles of FXI and FXII in coagulation and beyond
In addition to its procoagulant function in the activation of fIX, fXIa contributes to hemostasis by down-regulating fibrinolysis. This role is mediated by activation of thrombin-activatable fibrinolysis inhibitor (TAFI), also known as plasma carboxypeptidase U. TAFIa cleaves C-terminal lysines on fibrin, resulting in down-regulation of fibrinolysis by reducing the number of plasminogen and tissue-plasminogen-binding sites on fibrin. It makes intuitive sense, therefore, that the typical bleeding manifestations in fXI-deficient patients tend to be more pronounced at sites of enhanced fibrinolytic activity, such as in the nasopharyngeal or genitourinary tracts. Importantly, these patients do not experience the type of spontaneous intraarticular or intramuscular bleeding that is seen in patients with hemophilia. Factor XII also participates in fibrinolysis; fXIIa, via activation of pre-kallikrein, activates plasminogen and pro-urokinase, and thus has a seemingly paradoxical role in promoting fibrinolysis.9 This observation, together with the well-publicized demise of John Hageman from a pulmonary embolism in 196810 and several other case reports and series of thrombotic events in fXII-deficient subjects,11 led to a view that fXII deficiency is a hypercoagulable state in humans.12
In addition to their roles in coagulation, the contact factors collaborate to generate bradykinin from high-molecular-weight kininogen; bradykinin in turn mediates inflammatory responses by promoting vascular relaxation, neutrophil chemotaxis, and vascular permeability. Many of these effects are further augmented by fXIIa-mediated activation of the classic complement pathway (Figure 1). Recent animal studies have shown that not only fXI, but also fXII, may have a role in stabilizing platelet thrombi (reviewed by van Montfoort and Meijers in a separate chapter in this publication). In other words, separate from its formerly proposed role as an initiator of coagulation, fXII may have a newly discovered function in propagating thrombus formation in vivo. Conceptually, because fXII is apparently dispensable for normal hemostasis, this paradigm challenges McFarlane's notion that thrombosis is simply “hemostasis in the wrong place and at the wrong time.”13 It also raises the possibility of targeting factors XI and XII to prevent and/or treat certain types of thrombosis without incurring the same bleeding risk that is seen with current anticoagulants, including those that target fXa or thrombin. Preclinical data and early human trials addressing this novel concept will be addressed by Gailani in a separate chapter in this publication.

Protean roles of fXII and other contact factors in coagulation, inflammation, and fibrinolysis. HK indicates high-molecular-weight kininogen; and PK, prekallikrein. (Figure reprinted with permission from Gailani and Renné.37 )
Protean roles of fXII and other contact factors in coagulation, inflammation, and fibrinolysis. HK indicates high-molecular-weight kininogen; and PK, prekallikrein. (Figure reprinted with permission from Gailani and Renné.37 )
Relevance of animal data to humans
Although recent data generated in animal models are intriguing, the possibility exists that animal data fail to predict the situation in humans. For example, although fXII−/− animals were protected from thrombosis in experimental models of stroke and pulmonary embolus, fXII+/− (heterozygote) animals were not.14 This observation raises the possibility that intense pharmacologic inhibition of the contact factor(s) in question will be required to realize the full therapeutic benefit. It also suggests that, in population studies, the phenotype of interest may only be manifest in severely deficient subjects. This distinction between outcomes in severe and moderate deficiency is indeed illustrated in epidemiologic studies of survival in subjects with fXII deficiency. In a large Austrian cohort of almost 9000 individuals who were initially screened for thrombophilia, all-cause mortality on follow-up was analyzed according to fXII activity (fXII:c) levels. Individuals with >100% fXII:c had an overall survival that was similar to the Austrian population; however, mortality increased with stepwise decreases in fXII levels, such that those with fXII:c levels in the 10%–30% range had more than a 4-fold risk of death. Interestingly, however, the group with fXII:c levels <10% (n = 58) demonstrated no increased mortality, resulting in a U-shaped survival curve.15 The reason for the increased mortality in moderate-deficiency patients could not be determined because the relationship held true for noncancer mortality, all-cause vascular mortality, and mortality due to ischemic heart disease.
Furthermore, many factor deficiency states are clearly associated with the predicted outcome(s); for example, although fVIII deficiency leads to bleeding, excessively high fVIII levels are a risk factor for thrombosis. This is true regardless of the mutation causing the deficiency or excess. Conversely, the relationship between plasma levels and phenotype for some coagulation factor defects are much less predictable, as is the case with fibrinogen deficiency, in which severely reduced levels may be associated with thrombosis as well as bleeding.
The remainder of this chapter addresses those population studies that have focused on the relationship between fXI or fXII levels with venous thromboembolism (VTE), ischemic stroke (IS), or myocardial infarction (MI). These studies have included case-control and prospective cohort designs and are summarized in Tables 1 and 2, for fXII and fXI, respectively.
Epidemiologic studies examining associations between fXII and VTE, ischemic stroke, and MI/coronary heart disease
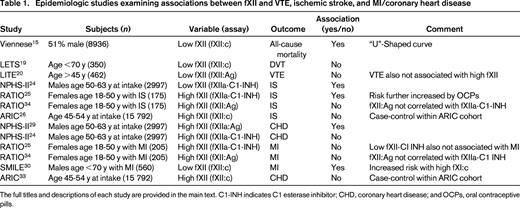
The full titles and descriptions of each study are provided in the main text. C1-INH indicates C1 esterase inhibitor; CHD, coronary heart disease; and OCPs, oral contraceptive pills.
Epidemiologic studies examining associations between fXI and VTE, ischemic stroke, and MI/coronary heart disease
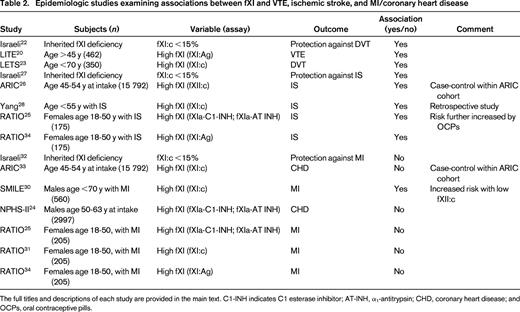
The full titles and descriptions of each study are provided in the main text. C1-INH indicates C1 esterase inhibitor; AT-INH, α1-antitrypsin; CHD, coronary heart disease; and OCPs, oral contraceptive pills.
VTE
FXII
John Hageman's death from pulmonary embolism occurred 12 days after he sustained pelvic fractures, during which time he was largely immobile.10 Although fXII deficiency per se was considered to be a risk factor for VTE,11 in retrospect, this conclusion was likely explained by reporting bias. Reevaluation of the reported cases16 and extended fXII-deficient pedigrees17 suggested that the majority of affected patients had other inherited or acquired risk factors that were more likely to explain the thrombotic events. In addition, it has been reported that a subset of antiphospholipid antibodies bind to fXII and may lead to an acquired “pseudo fXII deficiency”18 ; these antibodies, rather than the measured fXII deficiency, may therefore account for the observed thrombotic events.
In the Leiden Thrombophilia Study (LETS) involving 350 patients <70 years of age, there was no increased prevalence of fXII deficiency (fXII:c <57%) among patients with deep vein thrombosis (DVT) compared with controls. Importantly, there was also no variation in the risk across various severities of deficiency.19 The reverse situation, namely that high fXII levels are a risk factor for VTE (or indeed for thrombosis in general), has also not been borne out by epidemiologic investigation. In a nested case-control study from the Longitudinal Investigation of Thromboembolism (LITE) cohort, baseline (prethrombosis) fXII antigen (fXII:Ag) levels were measured in 462 adults (≥45 years of age) with incident VTE and in 1047 controls. The risk of VTE did not vary across the range of quintiles, indicating that neither fXII deficiency nor excess was associated.20 Of interest in this regard is the experience in patients with hereditary angioedema (HAE), a disorder in which C1 esterase inhibitor, one of the major physiological inhibitors of fXIIa, is deficient (type I or type II HAE) or fails to inhibit a mutated inactivation-resistant form of fXII (type III HAE).21 Despite the relative lack of regulation of fXII(a) in vivo, affected patients do not have any thrombotic propensity. Therefore, the most logical conclusion from the available data is that, whereas fXII deficiency (or indeed, excess) may not predispose to VTE, it also fails to be protective.
FXI
Unlike fXII, fXI deficiency—at least the more severe variant (<15% fXI:c)—has been reported to be protective against VTE.22 In a study of adult fXI-deficient patients in Israel, the rate of DVT (0/219) was statistically lower than the expected rate in the matched general population. Conversely, elevated fXI antigen levels were found to be a risk factor for VTE in the LITE study, with an odds ratio (OR) for VTE with fXI levels in the top quintile of 1.8 [95% confidence interval (CI), 1.3-2.7].20 In the LETS, the adjusted OR for DVT for subjects in the top decile was similar at 2.2 (95% CI, 1.5-3.2).23
IS
FXII
The relationship between fXII levels and IS in humans is unclear. Using an ELISA to measure fXIIa-C1 esterase inhibitor (fXIIa-C1-INH) levels in a nested case control study of middle-aged men in the Second Northwick Park Study (NPHS-II), lower levels of the complex were a risk factor for stroke.24 In contrast, in the Risk of Arterial Thrombosis In relation to Oral contraceptives (RATIO) study, which also measured fXIIa-C1-INH levels, elevated (>90th percentile) levels of the complex were associated with an increased risk of stroke, with an OR of 2.1 and a 95% CI of 1.3-3.5. Oral contraceptives increased the risk further.25 It is unclear whether this discrepancy was explained by sex or age differences or some other factor. However, when fXII:Ag levels were assayed in the same patients and controls, no association with stroke risk was apparent, suggesting that the analyte being measured likely accounted for the divergent conclusions.34 In the Atherosclerosis Risk in Communities (ARIC) Study, baseline fXII:c levels were measured in 89 incident stroke cases (42% male, 24% African American) and 406 random sample subjects.26 No difference between the mean values in the 2 groups could be discerned. As with VTE, reexamination of reported cases of fXII deficiency and thrombosis, as well as Swiss families with fXII deficiency, concluded that there was no association with stroke.16,17
FXI
The data linking fXI levels to stroke risk are somewhat more conclusive. First, as with VTE and using a similar approach in the Israeli population with fXI deficiency, Salomon et al concluded that fXI:c levels <15% incur protection against stroke.27 In the ARIC study, elevated levels of fXI:c were associated with an increased risk of IS (hazard ratio = 1.50, 95% CI, 1.10-2.05).26 In agreement with this result, Yang et al reported that fXI:c levels above the 95th percentile in a population <55 years of age were associated with an OR for stroke of 5.3.28 Furthermore, fXI:c and fXI:Ag levels were reasonably well correlated (R = 0.667). In the RATIO study, fXIa levels were measured by ELISA assays of the complexes of fXIa with its 2 principal inhibitors, C1-INH and α1-antitrypsin (AT-INH). In these younger women, levels of fXIa-C1-INH and fXIa-AT-INH complexes above the 90th percentile were associated with an elevated risk of stroke (OR = 2.8; 95% CI, 1.6-4.7 and OR = 2.3, 95% CI, 1.4-4.0, respectively).25 Unlike the case with fXII, the use of an antigen assay to measure plasma fXI levels (fXI:Ag) was in agreement with the conclusion using enzyme-inhibitor complexes.34 Therefore, the available data overall seem to support a link between high fXI levels and IS risk.
MI
FXII
In the NPHS-II study, higher baseline levels of fXIIa (measured by an ELISA specific for fXIIa) were predictive of coronary heart disease in middle-aged men.29 However, the opposite effect was seen in the same study when fXIIa-C1-INH levels were measured.24 In the RATIO study, neither higher nor lower levels of fXIIa-C1-INH levels were associated with MI in younger women.25 The same conclusion was reached when fXII:Ag levels were measured in these patients.34 In the Study of Myocardial Infarctions Leiden (SMILE) study, levels of fXII:c were evaluated as a risk factor for MI in 560 men and 646 controls younger than 70 with a first MI.30 Factor XII:c levels were lower in cases than in controls. Specifically, the adjusted OR of MI for men in the highest quintile compared with those in the lowest quintile was 0.4 (95% CI, 0.2-0.5). In a case-cohort sample from the ARIC study, fXII:c was measured in 368 incident cases and 412 random controls. No association was observed between fXII levels and coronary heart disease events, defined as hospitalized MI, a definite CHD death, proven silent MI, or a coronary vascularization.33
It is evident from these studies that a firm conclusion on the role of fXII as a risk factor for MI is not established. It is possible that the discrepant results are explained by chance or other factors such as study design or case definitions. However, the particular assay selected is also likely to be an important variable, with major differences sometimes apparent in studies measuring zymogen versus enzyme levels. Therefore, when measuring fXIIa or the fXIIa-C1-INH complex, the extent and/or rate of fXII activation could be quite heterogeneous among subjects. Therefore, levels of these complexes do not necessarily correlate with fXII:c or fXII:Ag levels and, indeed, recent data from the RATIO study have indicated that high fXII antigen levels do not correlate with activated protein-inhibitor complexes.34 Furthermore, as allued to previously, the consequences of moderate and severe fXII deficiency may be very different.15
FXI
In the Israeli fXI-deficient population, no protection from MI was noted, in contrast to VTE and stroke.32 In the prospective case-cohort study of ARIC participants, the association of fXI:c with coronary heart disease was not significant when adjusted for Framingham risk factors.33 In contrast, in the SMILE study, the risk of MI was increased for each quintile of fXI:c compared with the bottom quintile even after adjustment for cardiovascular risk factors and fXII:c levels.31 Interestingly, although fXI:c and fXII:c levels were correlated, the opposite effects on risk for MI were also synergistic; therefore, the highest risk was found in men with both high fXI:c and low fXII:c. To explain this seeming paradox, the investigators suggested that any effect of fXII on risk of MI may not be mediated via fXI, but rather through some other mechanism.30 In the NPHS-II study, plasma levels of factor XIa complexes (fXIa-C1-INH and fXIa-AT-INH) were not associated with a greater risk of coronary heart disease in middle-aged male participants.24 In agreement with this conclusion, the RATIO study demonstrated that elevated levels of fXIa complexes,25 fXI:c,31 and fXI:Ag34 were not associated with a greater risk of MI in young women. In contrast to these studies, cross-sectional studies in patients at presention with acute coronary syndrome have demonstrated elevated levels of fXIa activity,35 as well as fXIa-C1-INH and fXIa-AT-INH complexes.36 Overall, however, the evidence that FXI is a risk factor for MI is not as well established as for VTE or stroke. Whether this represents some unique difference in the coronary vasculature or in the role of the intrinsic pathway factors in MI remains to be established.
Conclusions
Although there is reasonable consistency in the data linking fXI deficiency or excess to hemostatic or thrombotic disorders, respectively, the same cannot be said for fXII. It remains unclear how many of the unresolved issues can be explained by assay methodology—which has included measures of fXII:c, fXII antigen, fXIIa antigen, and fXIIa-C1-INH levels in plasma—or some other variable such as the study population or design. Therefore, epidemiologic studies can offer only limited guidance to the ultimate decision of whether pharmacologic inhibition of fXI or fXII (or perhaps both) represents a more efficacious and safe antithrombotic approach. This information may only be obtained by carefully designed and monitored intervention studies in primates and in humans.
Acknowledgments
This work was supported by National Institutes of Health grant UO1HL117659 (to N.S.K.).
Disclosures
Conflict-of-interest disclosures: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Nigel S. Key, MB, ChB, FRCP, Department of Medicine, Division of Hematology/Oncology, University of North Carolina at Chapel Hill, 303 Mary Ellen Jones Building, CB #7035, Chapel Hill, NC 27516; Phone: (919)966-3311; Fax: (919)966-7639; e-mail: nigel_key@med.unc.edu.
