
Abstract
Hemophilia A and B are bleeding disorders that result from functional deficiencies in specific circulating blood clotting factors termed factor VIII (FVIII) and factor IX (FIX), respectively, and collectively display an incidence of 1 in 4000 male births. Stem cell transplantation therapies hold the promise of providing a cure for hemophilia, but currently available transplantable stem cell products do not confer endogenous FIX or FVIII biosynthesis. For this reason, stem cell–based approaches for hemophilia have focused primarily on genetic engineering of pluripotent or multipotent stem cells. While pluripotent stem cells have been branded with high expectation and promise, they remain poorly characterized in terms of clinical utility and safety. In contrast, adult-lineage-restricted stem cells are established agents in the clinical armamentarium. Of the clinically established stem cell types, hematopoietic stem cells (HSCs) are the most utilized and represent the standard of care for several genetic and acquired diseases. Furthermore, HSCs are ideal cellular vehicles for gene therapy applications because they self-renew, repopulate the entire blood lineage while concurrently amplifying the transgene copy number >106 fold, and also have direct access to the bloodstream. Current research on HSC transplantation gene therapy approaches for hemophilia A and B is focused on the following: (1) identification of safe and efficient methods of nucleic acid transfer, (2) optimization of transgene product expression, (3) minimization of conditioning-regimen-related toxicity while maintaining HSC engraftment, and (4) overcoming preexisting immunity. Based on the existing data and current rate of progress, clinical trials of HSC transplantation gene therapy for hemophilia are predicted to begin in the coming years.
Learning Objective
To gain an understanding of the key issues surrounding the implementation of stem cell therapies for hemophilia
Current state of hemophilia care
An ongoing revolution in the care of persons with hemophilia A and B has been taking place over the past half century. Today, routine perinatal diagnosis and implementation of life-long prophylaxis using factor VIII (FVIII) and factor IX (FIX) replacement products, respectively, for hemophilia A and hemophilia B, are reducing bleeding-related morbidity and restoring life expectancy. Recent and prominent examples of continued therapeutic advancement are the bioengineered “long-acting” FIX and FVIII products, as well as activated recombinant FVII and bispecific-antibody-based FVIII mimetics, for the treatment of FVIII inhibitors.1 However, none of these treatments is curative and significant deficiencies exist in global hemophilia care. For example, there remains polarizing disparity in the range of care available throughout the world, with ∼70% of persons with hemophilia not receiving any treatment and often not being specifically diagnosed. In the untreated state, severe hemophilia is a lethal disease with median mortality in the preteenage years and significant morbidity due to progressively debilitating joint arthropathy. The limited treatment availability is a consequence of factor product costs, which frequently exceed $250 000 per patient per year in the United States and are a lifelong expense. Although significant efforts are being directed toward economic treatment strategy optimization, it does not seem feasible that the current approach to disease management through factor-infusion-based prophylaxis is economically achievable for the majority of patients in the 21st century. For those who do receive treatment, negative aspects of factor infusion therapy remain, including the invasiveness of multiweekly, lifelong IV infusions of factor products, the need for central venous access devices, and immune responses to the infused product that render it ineffective in as high as 20%–30% of severe hemophilia A patients. Therefore, even though tremendous progress has been made in the treatment of hemophilia through research and development, the pursuit of novel and transformative therapeutic approaches remains an active area of investigation.
Rationale and clinical experiences of cell and gene transfer-based therapy of hemophilia
Several characteristics of hemophilia A and B make them amenable to stem cell- and gene transfer–based therapeutic strategies. First, small increases in circulating FVIII or FIX levels can render significant clinical benefit. For example, increasing the baseline FVIII level to >1% of normal, which represents a minimal plasma concentration of 10 pM (or 1 ng/mL), can eradicate spontaneous bleeding episodes, and achieving levels in the range of 500 pM (or 50 ng/mL) could completely cure the disease. Second, gene therapy should be more economical and less invasive than factor replacement therapy because it would consist of a single treatment event. Third, and specific to hemophilia A, FVIII, unlike FIX, does not depend on gamma-carboxylation for function, which is a cell type–specific activity and is most prevalent in hepatocytes. Therefore, FVIII can be biosynthesized and secreted into the bloodstream by most cell types with vascular access without loss of specific activity.
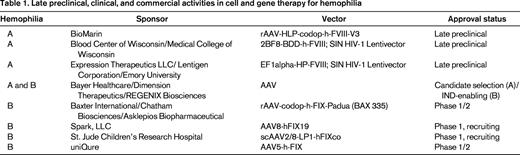
There have been 5 previous (now closed) and 3 ongoing clinical trials of various gene therapies for hemophilia.2 Each of the initial 5 trials was halted due to either immune responses to the vector or lack of evidence for therapeutic efficacy. Despite these setbacks, clinical gene therapy of hemophilia continues with 3 ongoing, independent trials (St. Jude Children's Research Center/University College London; Spark, LLC; and Baxter International/Chatham Therapeutics) all testing liver-directed adeno-associated viral (AAV) vectors encoding FIX for the treatment of hemophilia B. Of these trials, only initial results from the St. Jude trial have been reported.3 In early 2014, BioMarin Pharmaceutical released a statement indicating their intent to initiate a similar trial for hemophilia A, which is the 5-fold more prevalent form of the disease. Most recently, in June 2014, Bayer HealthCare agreed to a collaboration with Dimension Therapeutics to commercialize another independent AAV gene therapy product for hemophilia A. As these activities clearly indicate, gene therapy of hemophilia is being aggressively pursued and multiple commercial entities have active preclinical and clinical hemophilia gene therapy pipeline programs (Table 1).
Stem cells as therapeutics
One anticipated shortcoming of AAV-based gene therapy approaches is limited therapeutic duration due to the turnover of the targeted cell types (eg, hepatocytes). In contrast, targeting stem cells for genetic modification and delivery of FVIII or FIX theoretically presents as the most durable approach. In the simplest sense, stem cells can be defined as undifferentiated cells that possess 2 key properties, self-renewal and potency (ie, cellular differentiation potential). From a clinical perspective, these properties endow stem cells with the ability to not only treat, but also to cure a multitude of human diseases, which can include hemophilia. There are 2 broad categories of stem cell types being investigated for clinical application. The first are the pluripotent stem cell (PSC) types that are naturally present during development. These include embryonic stem cells (ESCs) derived from the inner cell mass of a blastocyst, embryonic germ cells (EGCs) isolated from the fetal gonads, and induced PSCs (iPSCs) that are engineered from differentiated cells. The second are the multipotent adult stem cells found in many organs, which include BM-derived hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs). Currently, our understanding of the clinical translatability of ESCs, EGCs, and iPSCs remains the least mature because their safety, manufacturing, and efficacy have yet to be rigorously established and significant scientific challenges remain. However, despite these deficiencies, several clinical trials using PSCs are ongoing.4 In contrast, HSC transplantation (HSCT) is and has been the standard of care for the treatment of a range of monogenic diseases and several types of cancer for more than half a century, even though certain aspects of the HSC phenotype/identity remain elusive. Furthermore, HSCs have proven to be excellent targets for genetic manipulation and ongoing clinical trials continue to demonstrate the utility of targeting this cell population as a means of delivering nucleic acid therapeutics for the treatment and cure of several previously intractable diseases.
Origins and limitations of FVIII and FIX biosynthesis
For many monogenic blood diseases, allogeneic stem cell transplantation from unaffected donors is a successful approach to disease management. However, a prerequisite of this approach is the endogenous expression of the deficient protein by the donor cells. It has been known for decades that orthotopic liver transplantation cures hemophilia in dogs and humans. This is not unexpected given that vitamin K–dependent coagulation factors such as FIX require the activities of 2 proteins highly expressed in hepatocytes, gamma-glutamyl carboxylase and vitamin K epoxide reductase. However, several paradoxical observations surrounding FVIII biology made its cellular biosynthetic source more ambiguous. First, it was known that FVIII levels increase during fulminant hepatic failure, whereas the levels of all other coagulation factors decrease. Second, it was observed clinically that FVIII levels did not decrease in the recipient of a liver from a donor with mild hemophilia.5 For these reasons, it has been widely speculated that both hepatic and extrahepatic sources of FVIII exist, a role likely held by endothelial cells of both the liver sinusoids and elsewhere. However, it was not until early 2014 that endothelial cells clearly were identified as the primary endogenous source of FVIII by the Montgomery and Ginsburg laboratories using distinct murine genetic engineering approaches involving cell type–specific F8 and Lman1 gene conditional knockout technology, respectively.6,7 Concurrently, Jacquemin et al came to a similar conclusion by studying primary human liver sinusoidal endothelial cell and hepatocytes. They observed that endothelial cells, but not hepatocytes, contained measurable levels of FVIII coagulant activity.8 Before these studies, there existed conflicting reports and significant speculation that endothelial cells, hepatocytes, and hematopoietic and mesenchymal cells were important contributors to the circulating pool of FVIII. Without clinical evidence of nonendothelial FVIII biosynthesis and nonhepatocyte FIX biosynthesis and with no existing source/transplantation protocols for endothelial or hepatocyte stem cells, allogeneic transplantation does not appear to be a viable near-term option. Therefore, in the near future, stem cell–based therapy strategies for hemophilia A and B likely will remain dependent on gene transfer technology.
During the research and development of recombinant FVIII products, Kaufman et al observed that human FVIII (h-FVIII) is secreted at levels 2–3 orders of magnitude lower than other plasma proteins in heterologous expression systems.9 For many secreted proteins, transit from the endoplasmic reticulum to the Golgi apparatus is rate limiting and the primary determinants of this limitation are believed to be specific amino acid sequences within the molecule itself.10,11 Using this knowledge, we and others have focused on the development of biobetter FVIII molecules and their implementation into gene transfer vectors that can be used in stem cell gene therapy applications. For example, it was shown by Kaufman et al that coexpression of von Willebrand factor results in the improved accumulation of recombinant FVIII in the culture medium of heterologous cells.12 Pipe et al demonstrated that restoration of several N-linked glycosylation sites from the deleted B domain of a B domain deleted (BDD) human (h)-FVIII construct facilitated increased interaction with mannose-binding lectin protein-1 (LMAN1), resulting in more efficient carbohydrate-facilitated transport from the endoplasmic reticulum to the Golgi apparatus.13 Nathwani et al combined a similar approach with codon optimization and demonstrated significant improvement in FVIII production from liver-directed AAV gene transfer systems.14 In addition, we have shown that BDD porcine FVIII (p-FVIII) is expressed at 10- to 100-fold greater levels than BDD-h-FVIII in vitro.15,16 Furthermore, we identified sequences within p-FVIII that are necessary and sufficient for the greater expression,11 demonstrated that this differential occurs through reduced engagement of the unfolded protein response pathway,10 and have used this information to generate humanized, human/porcine (HP) high-expression FVIII constructs that retain this biosynthetic advantage.17,18 A similar strategy is being pursued in gene therapy applications for hemophilia B through the use of a naturally occurring human FIX variant termed FIX-Padua (R338L) that exhibits greater specific procoagulant activity than wild-type human FIX. Inclusion of this modified FIX has been shown to improve vector and transgene potency in preclinical AAV gene therapy studies.19,20 Collectively, evidence is building to support FVIII and FIX transgene engineering approaches as critical components to the success of stem cell–based gene therapies for hemophilia A and B. However, the safety and efficacy of these molecules remain to be tested and proven in clinical trials.
PSC-based therapies
The first clinical trial of a PSC-derived product was initiated by Geron in 2009 for acute spinal injury and involved a human ESC (hESC)–derived oligodendrocyte product. In 2014, there are 7 ongoing clinical trials of hESC-derived products and all except 1 target macular degeneration (acute wet age-related, Stargard's, and advanced dry age) and are sponsored by Advanced Cell Technology. The remaining trial, sponsored by Assistance Publique–Hopitaux de Paris, involves the testing of CD15+ Isl-1+ hESC-derived progenitors in ischemic heart disease. At present, no ongoing clinical PSC approaches appear prototypical for application to hemophilia, but more research and development in this area appear warranted. Toward this goal, several research groups recently reported on the feasibility of PSC therapy for hemophilia. For example, Xu et al generated iPSCs from tail tip fibroblasts of healthy mice, differentiated the cells toward an endothelial phenotype (CD31+ CD34+ Flk-1+), and then transplanted them directly into the livers of irradiated hemophilia A mice.21 After transplantation, recipient mice displayed plasma FVIII activity levels in the range of 8%–12% and survived a bleeding challenge via tail transection assay. Ohmori et al also investigated the therapeutic potential of murine iPSCs, although SIV-based lentivector gene transfer of a BDD h-FVIII transgene was used to enhance, if not provide the sole source of, FVIII.22 FVIII production was demonstrated in vitro from undifferentiated genetically modified iPSCs and teratomas formed in nude mice. Following up on these findings, Oshimura et al genetically modified iPSCs derived from embryonic fibroblasts of hemophilia A mice using human artificial chromosomes encoding 1-4 copies of an h-FVIII transgene under the control of a platelet factor 4 promoter.23 Genetically modified iPSCs were differentiated into megakaryocytes/platelets in vitro and h-FVIII mRNA expression was detected in the resulting cells. Overall, these studies represent early-stage, in vitro, and preclinical animal proof of concept that require validation, safety testing, and product refinement for clinical translation.
Multipotent adult stem cell therapies: MSCs
Two adult stem cell populations residing in the BM compartment, MSCs and HSCs, may hold therapeutic promise for hemophilia. MSCs are an ideal cellular vehicle for the delivery of nucleic acid–based therapeutics due to their ease of harvest, robust cell processing potential, mesenchymal lineage potency, and efficiency of stable gene transfer.24 Although MSC cultures are typically heterogeneous in nature, under the strictest definition, they are identified and characterized by their differentiation potential into fibroblast CFU, osteoblasts, chondrocytes, and adipocytes. However, because MSCs have not been demonstrated in vivo to regenerate or maintain a tissue compartment and thus do not meet the strict definition of a stem cell, they also can be termed BM-derived stromal cells. Currently, MSCs are the subject of ∼500 clinical trials for a wide array of medical conditions including, but not limited to, osteogenesis imperfecta, Crohn's disease and ulcerative colitis, multiple sclerosis, diabetes, liver cirrhosis, and knee cartilage injury, which as suggested by Ebihara et al, may be applicable to hemophilic joint arthropathy.25
Because cells of mesenchymal origin are not believed to be significant contributors to circulating FVIII or FIX levels, gene transfer appears to be a necessary component for the clinical utilization of MSCs toward correction of bleeding in hemophilia. Several groups of investigators are pursuing this approach and, of these, one particularly promising but preliminary study was conducted in an ovine model of hemophilia A.26 In that study, Porada et al used adult BM-derived MSCs isolated from paternal donors that were genetically modified using HIV-based lentivector encoding a BDD p-FVIII transgene. Genetically modified MSCs were transplanted into the peritoneal cavity of 2 affected hemophilia A recipients, the first of which already harbored a low-titer inhibitor against h-FVIIII and received a dose of 3 × 107 cells. The second did not yet possess an inhibitor and received an initial dose of 1.2 × 108 cells. Before transplantation, both animals presented with bleeding episodes and progressive morbidity despite receiving on-demand therapy with h-FVIII products. After stem cell therapy, chronic joint arthropathy resolved in both animals and they remained bleeding episode free for >2 months in the absence of FVIII infusions. Both animals developed high-titer anti-p-FVIII inhibitors that cross-reacted with and negated the efficacy of h-FVIII products. This was not unexpected because we had observed similar results in hemophilia A mice transplanted with genetically modified MSCs expressing BDD p-FVIII.27 These results highlight the immunogenic potential of MSC (and other stem cell)-delivered FVIII molecules. Despite this immune reaction, the animals did not bleed for an extended period of time. It was speculated that the phenotypic correction resulted from the widespread distribution of genetically modified MSCs that likely homed to sites of joint injury and released a local pool of FVIII, thereby providing procoagulant function before neutralization. Unfortunately, both animals eventually succumbed to bleeding-related deaths caused by severe trauma, which in one instance was ingestion of a sharp object and the other was aggressive behavior–related injury. Notwithstanding their premature deaths, these hemophilic sheep represent impressive case studies that may predict the clinical utility of genetically modified MSCs in hemophilia both with and without preexisting FVIII immunity.
A current challenge to the development of MSC-based therapeutics are poor pharmacodynamics and pharmacokinetics. To address these obstacles, Hortelano et al have been investigating the ability of different biomaterials to improve cord blood–derived MSC viability and FIX production after transplantation.28 They have shown that both arginine-glycine-aspartate (RGD) and fibrinogen-coated alginate-PLL microcapsules improve cell survival over noncoated microcapsules. However, only fibrinogen-coated microcapsules improved FIX secretion. Similarly, Galipeau et al demonstrated that 3D scaffolds composed of a hydroxyapatite–PLGA composite and coated with collagen supported long-term engraftment of genetically modified MSCs and systemic delivery of FIX in hemophilia B mice.29 These studies are advancing rapidly and the data support the continued investigation of MSCs as therapeutic agents for hemophilia.
Multipotent adult stem cell therapies: HSCs
HSCs are an ideal target for life-long expression of therapeutic proteins because BM transplantation studies in children have shown that transplanted HSCs survive for the lifetime of the recipient. However, because HSCs are extremely rare and typically quiescent, genetic engineering of this cell population has been difficult. For example, methods to introduce nucleic acids by routine laboratory methods such as electroporation or other plasmid-based transfer systems have been largely unsuccessful. In contrast, the use of γ-retroviral vectors for gene transfer showed early promising results. Based on a multitude of studies, the field now considers the use of HIV-based lentivectors as the most efficient method of introducing new genetic material into HSCs. The first reason for this is that lentivectors have the ability to insert their genetic sequence into the nuclear chromosomes of target cells. Once the sequence is integrated into the genome, host cellular machinery transcribes the proviral sequence. Second, HSCs can durably reconstitute the entire hematopoietic compartment, including myeloid, lymphoid, megakaryocytic, and erythroid lineages, through both self-renewal and cellular differentiation. Importantly, this aspect of HSC biology provides a natural method for transgene amplification. For example, engraftment levels of 1% genetically modified cells after HSCT can result in as many as 2 × 108 WBCs, 2 × 1011 RBCs, and 1012 platelets containing the transgene and theoretically producing the protein product. Although recombinant lentivectors are among the most well-studied gene transfer technologies and are being used in several ongoing clinical trials, there is the theoretical potential for insertional mutagenesis, a topic that has been well reviewed by others.30 Third, the HSC compartment is mostly quiescent and is not efficiently transduced by γ-retroviruses. However, unlike γ-retroviruses, lentivectors efficiently transduce quiescent cell populations. Finally, a particular benefit of using lentivector-transduced HSCs is that HSCT has been shown to facilitate the development of tolerance to neoantigens through establishment of mixed chimerism.31,32 This tolerogenic aspect of HSCT also encompasses autologous HSCs that have been genetically modified to express a protein that previously was absent or nonfunctional,32 such as FVIII in patients with hemophilia A. This phenomenon was originally dubbed “molecular chimerism.” We now modify it slightly for clarity in the context of HSCT gene therapy of hemophilia A and refer to it as “unimolecular microchimerism” to reflect the presence of a single new molecular antigen (ie, FVIII) and the low-level (micro) chimerism that is achievable using reduced toxicity conditioning regimens. Immune nonresponsiveness or even tolerance induction is a critical requirement of cell and gene therapy applications in which the immune system can be a major barrier to therapeutic success. Current preclinical research in the field of HSCT gene therapy of hemophilia is focused on 4 main issues. The first, as mentioned earlier, is the general understanding of lentivector integration profiles and risk of insertional mutagenesis.30 The second is the identification of the optimal hematopoietic cell types for expression of either FVIII or FIX. The third is the development of protocols and agents designed to minimize transplantation conditioning regimen-related toxicity while maintaining sufficient and durable HSC engraftment. The fourth is overcoming the hurdle of preexisting immunity to FVIII or FIX. These aspects of HSCT gene therapy for hemophilia are the focus of the remainder of this chapter.
Evans and Morgan conducted the first preclinical study of HSCT-based gene therapy of hemophilia A.33 Although correction of the FVIII deficiency was not achieved, the study did provide evidence for immune tolerance, or at least nonresponsiveness, induction. Furthermore, it demonstrated the difficulties associated with the development of gene therapy applications for hemophilia A by the inefficiency of FVIII biosynthesis. The first preclinical HSCT gene therapy study to achieve sustained correction of FVIII activity to therapeutic levels was conducted by Hawley et al,34 who used a bicistronic vector construct incorporating both a BDD h-FVIII transgene and EGFP within γ-retroviral vector particles. This enabled the identification and enrichment of genetically modified stem and progenitor cells before transplantation, which thereby facilitated optimal transplantation of 100% genetically modified cells. Around this same time, Sakata et al showed that genetic modification of CD34+ cells using an SIV vector followed by transplantation into NOD/SCID mice resulted in detectable, albeit low, plasma h-FVIII levels.35 These studies highlighted several key findings. First, FVIII transgenes can be transferred by recombinant retroviral viral vectors without rearrangement issues. Second, sustained FVIII expression is achievable through HSCT into conditioned recipients. Third, however, these studies also identified that inefficient FVIII expression/biosynthesis is a translational barrier to HSCT gene therapy for hemophilia A.
Several strategies have been used to enhance FVIII expression, of which several successful ones involve genetic engineering of the h-FVIII transgene sequence. For example, we and others have shown that BDD p-FVIII is expressed at 10-fold or greater levels compared with BDD h-FVIII.11,15,16 This led to the rationale of using a recombinant murine stem cell (γ-retrovirus) viral vector encoding BDD p-FVIII in HSCT gene therapy studies. These studies were highly successful with respect to the plasma FVIII activity levels achieved in transplanted hemophilia A mice conditioned with lethal or sublethal irradiation-based conditioning.27 Not only were high levels of circulating FVIII activity achieved (>300% normal levels), but tolerance induction to both recombinant porcine and human FVIII was also shown.36 These successes led to a subsequent study in which we identified a nonmyeloablative transplant regimen consisting of busulfan and antithymocyte serum that facilitated sustained transgene expression and tolerance induction at low transgene copy number.37 Furthermore, the residues necessary for the high-level expression of BDD p-FVIII were confirmed and cloned into BDD h-FVIII, thereby generating a high-expression HP (91% human and 9% porcine) chimera. This bioengineered FVIII transgene demonstrated equivalent transgene product expression to BDD p-FVIII in vitro and in vivo using both SIV- and HIV-based expression vectors.11,17,18
An alternative, but also successful, preclinical HSCT gene therapy strategy incorporating nonengineered BDD h-FVIII, and more recently FIX, transgenes involves specific targeting of transgene expression to the megakaryocyte/platelet hematopoietic lineage. This approach was first demonstrated by Poncz et al using transplantation of HSCs from transgenic mice to correct the hemophilia A bleeding phenotype.38 The development of self-inactivating (SIN) lentivectors, which require internal promoters to drive constitutive expression or, alternatively, direct cell type–specific expression (eg, megakaryocyte/platelet) facilitated the translation of this approach to HSCT gene therapy. For example, the Shi and Montgomery laboratories have used a 0.89 kb ITGA2B (integrin αIIb) promoter to drive BDD-h-FVIII or h-FIX expression in murine models of hemophilia A and B, respectively. Similar success was achieved despite apparent reduced specific activity of the platelet-directed FIX, predictably due to incomplete gamma-carboxylation.39,40 Using platelet-directed expression vector designs, FVIII or FIX is expressed in megakaryocytes and packaged into platelets, where it is sequestered in granules until the platelets are activated at site(s) of hemostatic need (eg, vessel wall injury) and the granule contents are released into the bloodstream. Intracellular sequestration does make it more difficult to assess FVIII (or FIX) expression/biosynthesis and predict the degree of hemostatic protection available. However, as the Poncz and Montgomery groups have shown clearly, this treatment protects hemophilia A and B mice in tail-clip bleeding assays despite the lack of circulating FVIII or FIX activity, respectively. In 2014, the first large animal (canine hemophilia A) trial of HSCT gene therapy was reported by Wilcox et al.41 In total, 3 animals were studied, 2 of which received autologous mobilized peripheral blood cells transduced with lentivector encoding the identical 0.89 kb ITGA2B promoter driving BDD h-FVIII. The third animal received cells transduced with lentivector encoding a shorter 0.67 kb fragment of the ITGA2B promoter driving a hybrid BBD-h-FVIII/von Willebrand factor propeptide signal and D2 domain designed to traffic FVIII to the α-granule. After transplantation, 2 of the 3 animals (2nd and 3rd) were bleeding episode free for 2.5 years without receiving canine FVIII infusions. Because differences in HSC identification and biology among mammalian species are known but not well understood, a late-stage preclinical testing requirement for investigational HSCT gene therapy products is transduction and xenotransplantation of human HSCs into immunocompromised mice. Toward this goal, Shi and Montgomery recently reported phenotypic correction of genetically immunocompromised hemophilia A mice after transplantation of genetically modified human cord blood CD34+ cells that had been transduced with the recombinant lentivector encoding BDD-h-FVIII driven by the 0.89 kb ITGA2B promoter.42 As highlighted by these newly published results, HSCT gene therapy approaches for hemophilia are progressing through the final stages of preclinical testing.
Another intriguing aspect of HSCT gene therapy is the ability to correct the bleeding phenotype and, in some protocols, eradicate inhibitors in the setting of preexisting anti-FVIII immunity. Aside from the 70% of persons with hemophilia who do not have access to treatment, patients with inhibitors to h-FVIII products are at the greatest risk of bleeding-related morbidity and premature mortality. Therefore, this population presents with the greatest need for products such as HSCT gene therapy. However, the additional challenges posed by preexisting immunity have precluded these patients from previous clinical gene therapy trials. To address this unmet clinical need, both our group and that of Shi and Montgomery have studied HSCT gene therapy in hemophilia A mice with preexisting immunity to h-FVIII and shown preclinical efficacy. Furthermore, both groups have identified the need for more intensive conditioning regimens to achieve similar levels of engraftment in the preimmunized setting as can be achieved with reduced-intensity conditioning in naive animals.36,43,44 For example, both groups showed the requirement for either high-dose total body irradiation or chemotherapy plus immune suppression using antithymocyte globulin to facilitate engraftment and efficacy. We also demonstrated that inclusion of the BDD p-FVIII transgene in conjunction with appropriate conditioning protocols not only restored curative plasma FVIII levels, but also permanently eradicated the existing FVIII inhibitors.36,43 However, at present, the consensus remains that testing of HSCT gene therapy in inhibitor patients should not be the first indication because, without the utility of FVIII replacement products, it may not be possible to maintain hemostasis throughout the transplantation procedure and follow-up period. However, we and others are continuing to investigate novel conditioning (eg, increased HSC specificity and toxicity) and immune-suppressing (eg, T- and B-cell blocking) agents with predictably lower overall toxicity profiles (eg, no induced thrombocytopenia) that may finally open the door for the testing of stem cell gene transfer therapies designed to address the unmet need of hemophilia patients harboring anti-FVIII or anti-FIX inhibitors.
Current outlook for clinical HSCT gene therapy
Translational aspects of HSCT gene therapy clinical product development, such as safety and manufacturing, are coming to the forefront and are beginning to be addressed. For example, an emerging realization is that each lentivector is unique in terms of insertional mutagenesis risk, manufacturing yield, and potency. For example, in GMP manufacturing studies performed in collaboration with Lentigen Corporation, we have observed striking (>10-fold) differences in vector manufacturing yield depending on the internal promoter incorporated (data not shown). Strong promoters, such as elongation factor 1 α (EF1α), may be competing with the promoter driving viral genome production (ie, the RNA molecule that is packaged into the recombinant viral particles), thus decreasing the number of fully packaged viral particles produced during a manufacturing run. However, if the internal promoter is “tuned” with the viral promoter, high-titer virus can be produced at clinical scale. One way we have been able to tune the vector cassette is to use an internal promoter that either is weaker than the promoter driving vector genome production or is not active in packaging cells but is active in targeted hematopoietic lineages (eg, monocytes/macrophages). Finally, we and others have observed a decrease in vector potency in the murine hemophilia A model as we have transitioned to safer vectors (eg, from murine stem cell viral vectors using viral long terminal repeat promoters, to SIN lentivectors incorporating internal long terminal repeat promoters, to similar vectors containing endogenous constitutive promoters, and finally to SIN lentivectors incorporating cell type–specific promoters) and conditioning regimens.
Concluding remarks
Current hemophilia treatment using factor replacement products is restricted by access, cost, invasiveness, duration, and immunogenicity. Therefore, a curative stem cell product would be transformative. Hemophilia A has many characteristics that make it amenable to HSCT-based gene therapy, such as (1) a low therapeutic threshold, (2) a large therapeutic window, (3) no known target cell type limitations other than access to the bloodstream, and (4) an adequate patient population for clinical testing. However, it also presents challenges such as the biosynthetic inefficiency and potent immunogenicity of h-FVIII compared with other plasma proteins. Fortunately, progress toward improving the biosynthetic efficiency through transgene/protein bioengineering and reducing the risk of immunogenicity through transplantation conditioning regimens is being achieved. Currently, HSCT gene therapy strategies incorporating HIV-1–based lentivectors encoding FVIII or FIX under the control of either constitutive or cell type–specific promoters are rapidly advancing from preclinical to clinical testing and may one day be applied successfully to the inhibitor setting. Successful implementation of a stem cell–based cure for hemophilia through HSCT gene therapy hinges on several key factors including improving gene transfer efficiency and transgene product expression; avoidance of immune responses to the vector, genetically modified cells, and transgene product; and obtaining adequate stem cell engraftment while maintaining a favorable risk/benefit ratio. For hemophilia A, at least one program/approach has advanced to the stage of Food and Drug Administration (FDA) review. Specifically, a proposed clinical trial of HSCT gene therapy incorporating an SIN lentivector encoding HP-FVIII has been reviewed by the Recombinant DNA Advisory Committee as well as the FDA in a pre-Investigational New Drug meeting and funding for this trial has been obtained through the National Institutes of Health and Hemophilia of Georgia. Therefore, it is anticipated that one, if not several, approaches using HSCT gene therapy of hemophilia A will enter clinical trials in the coming years.
Acknowledgments
This work was supported by the National Institutes of Health (Grants HL112309, HL092179, and HL114241 to C.B.D. and H.T.S. and Grant HL115171 to C.B.D.) and Hemophilia of Georgia (H.T.S.). The authors thank Shannon Meeks for thoughtfully reviewing the manuscript.
Disclosures
Conflict-of-interest disclosures: C.B.D. and H.T.S. are cofounders and hold equity interests in Expression Therapeutics, which owns the intellectual property associated with HP-FVIII (ET3). The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. C.B.D. has consulted for Bayer HealthCare. Off-label drug use: None disclosed.
Correspondence
Christopher B. Doering, PhD, 2015 Uppergate Drive, Emory Children's Center, Room 450, Atlanta, Georgia 30322; Phone: (404)727-7988; Fax: (404)727-4455; e-mail: cdoerin@emory.edu.
