
Abstract
Common variable immunodeficiency (CVID) is a rare immune deficiency characterized by low levels of serum IgG, IgA, and/or IgM, with a loss of Ab production. The diagnosis is most commonly made in adults between the ages of 20 and 40 years, but both children and much older adults can be found to have this immune defect. The range of clinical manifestations is broad, including acute and chronic infections, inflammatory and autoimmune diseases, and an increased incidence of cancer and lymphoma. For all of these reasons, the disease phenotype is both heterogeneous and complex. In the past few years, data from large patient registries have revealed that both selected laboratory markers and clinical phenotyping may aid in separating groups of subjects into biologically relevant categories. CVID consists of 2 phenotypes, 1 in which infections are the characteristic and another in which impressive inflammatory and/or hematologic complications also develop, including lymphadenopathy, splenomegaly, autoimmune cytopenias, enteropathy, and/or and granulomatous disease. These phenotypes appear to be stable, are related to immunologic and inflammatory markers, and are predictive of outcomes. This review outlines current understanding about this syndrome based on studies of large cohorts, highlighting the evaluation and treatment of complications and, in particular, the autoimmune and inflammatory conditions that affect these patients.
Introduction
Common variable immune deficiency (CVID) is considered to be a collection of primary immune deficiency diseases characterized by reduced serum levels of IgG, IgA, and/or IgM, with reduced or absent specific Ab production.1 The serum IgG is always reduced and is generally less than 400 mg/dL. Due to the relative prevalence (1 in 25 000 to 1 in 50 000) and numbers of medical encounters, CVID is a clinically important immune defect.2,3 Most patients are diagnosed between the ages of 20 and 40 years, but approximately 20% are under the age of 20.3 Generally, we are reluctant to diagnosis CVID in young children (under age 4) because physiologic immaturity can mimic CVID in the early years; also, this period of delay allows other diagnostic considerations to be explored. Unfortunately, most internists are not familiar with CVID, perhaps because primary immune defects are more commonly understood to be pediatric ailments. Added to this view is that is that the symptoms of CVID can be so heterogeneous, causing the patient to come first to medical attention in medical specialties of otolaryngology, respiratory medicine, gastroenterology, rheumatology, or others. Somewhat confusingly, a proportion of patients (perhaps 5%) do not have a significant number of infections, but instead come to medical attention due to the onset of selected inflammatory or autoimmune complications characteristic of CVID. All of these factors together contribute to an average delay of 6-7 years in the diagnosis of this syndrome; in many cases, characteristic symptoms preceded the diagnosis by an additional number of years.2–6
As might be expected when there is a lack of protective Ab, sinopulmonary tract infections are the main symptoms for most CVID subjects. Approximately half have had at least one episode of pneumonia, and others have had so many episodes that they have lost count. When there is no history of pneumonia, patients generally have recurrent bronchitis, sinusitis, and or otitis. The standard of care in CVID is replacement Ig given at frequent intervals for life. This can be given either by the IV or the subcutaneous route because both are equally effective. The doses range from 300 mg/kg every 3 weeks to 600 mg/kg a month IV or prorated doses for subcutaneous delivery. The choice of the route is best dictated by patient choice and convenience. However, although Ig therapy greatly reduces the number of bacterial infections7 and likely enhances survival,2 it does not appear to address the characteristic inflammatory complications consisting of progressive lung disease, gastrointestinal and granulomatous disease, autoimmunity, lymphoid hyperplasia and infiltrative disease, and the development of cancer, especially lymphoma. These complications now appear to be the major cause of morbidity and death in CVID.1,2,6 Treatment options and outcomes of these inflammatory and autoimmune complications are discussed herein.
Inflammatory and autoimmune complications
One of the interesting facets of CVID is that patients can quite often be classified into 2 main groups by disease phenotypes that are apparently stable over time. These 2 groups contain subjects with a history of infections and those who may have infections, but in addition, a variety of inflammatory and/or autoimmune conditions. For the latter set of patients, these medical problems have emerged as the manifestations of CVID that are most difficult to handle because one or more forms of immune suppression may be needed. This categorization becomes apparent when studying the large collections of patients assembled in Europe and in the United States. In the largest European study, 334 subjects from the European Society for Immune Deficiency (ESID) were followed for an average of 25.5 years; 71% had one or more of the inflammatory/autoimmune complications and the reminder had infections only.2 In the US group, 476 patients studied from one medical center, 68% had one or more of these manifestations and the rest had infections as the only manifestation.6
In the US group, the most prevalent of these conditions, even while on Ig therapy, was progressive lung disease in 28.5%, conceivably a residual of longstanding infections. However, in these cases, lymphocytic and/or granulomatous pulmonary infiltrates led to lung failure, suggesting an aggressive inflammatory component. More difficult to understand are the hematologic and organ-specific autoimmune diseases that were diagnosed in 12%-46% of subjects in the ESID report,2 in 28.6% of the US subjects,6 and in up to 20% of a cohort examined in France.8 As in all previous studies, for unclear reasons, the most common autoimmune diseases in CVID were immune thrombocytopenia followed by autoimmune hemolytic anemia. Other autoimmune conditions in the US study included anticardiolipin Ab, antiphospholipid syndrome, diabetes mellitus, inflammatory bowel disease, pernicious anemia, rheumatoid arthritis, juvenile rheumatoid arthritis, uveitis, multiple sclerosis, neutropenia, primary biliary cirrhosis, systemic lupus erythematosus, autoimmune thyroid disease, vasculitis, psoriasis, and vitiligo (Table 1 and Figure 1).6
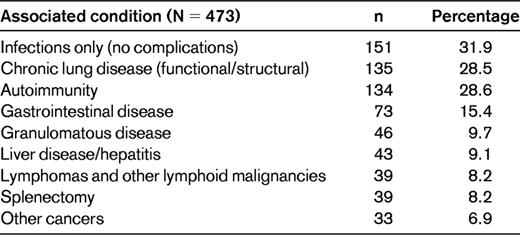

Autoimmunity in CVID. Of 473 patients with CVID, 134 (28.6%) had autoimmunity.6 The main autoimmune diseases are shown here. In the category “other” are included neutropenia, pernicious anemia, anticardiolipin Ab, antiphospholipid syndrome, diabetes mellitus, juvenile rheumatoid arthritis, uveitis, multiple sclerosis, systemic lupus erythematosus, autoimmune thyroid disease, lichen planus, vasculitis, vitiligo, and psoriasis. ITP indicates immune thrombocytopenia; AIHA, autoimmune hemolytic anemia; RA, rheumatoid arthritis.
Autoimmunity in CVID. Of 473 patients with CVID, 134 (28.6%) had autoimmunity.6 The main autoimmune diseases are shown here. In the category “other” are included neutropenia, pernicious anemia, anticardiolipin Ab, antiphospholipid syndrome, diabetes mellitus, juvenile rheumatoid arthritis, uveitis, multiple sclerosis, systemic lupus erythematosus, autoimmune thyroid disease, lichen planus, vasculitis, vitiligo, and psoriasis. ITP indicates immune thrombocytopenia; AIHA, autoimmune hemolytic anemia; RA, rheumatoid arthritis.
Striking lymphadenopathy, usually cervical, mediastinal, or abdominal, is another common finding in CVID; however, as noted for the pulmonary infiltrates, the generalized expansion of the lymphoid compartment can include extension into other tissues such as the liver or gastrointestinal tract, leading to lymphoid collections in these organs. The incidence of splenomegaly is approximately 30% in most series2 ; it may be massive in some cases, which appears to be a stable condition. However, due to the enlarged spleen or hypersplenism, but more probably because of uncontrolled autoimmunity (ie, immune thrombocytopenia or autoimmune hemolytic anemia), a significant proportion of CVID patients have had a splenectomy. Since its introduction, the use of rituximab has greatly reduced the likelihood of splenectomy for autoimmunity alone.9 Lymphoid tissues may also contained noncaseating granuloma, suggesting sarcoid-like changes. Noncaseating granulomatous disease, similar to sarcoidosis, has an incidence of 8%-20%, depending on the populations studied.10,11 In most cases, these are found in the lung, lymph nodes, liver, and skin, but any tissue can be involved. Organisms are always sought, but almost never found, suggesting that this is an unusual form of an inflammatory reaction. For unclear reasons, CVID subjects with granulomatous disease are more likely to develop autoimmunity, suggesting a related pathogenesis.
Gastrointestinal disease
The main gastrointestinal symptom in CVID is transient or persistent diarrhea.12,13 This is likely to be infectious, and Giardia lamblia is the most common organism, but other pathogens are not uncommon.14 Helicobacter pylori infection has been associated with gastritis.15 Inflammatory bowel disease remains a significant problem in 19%-32%,13,16 leading to chronic, even severe diarrhea characterized by weight loss, steatorrhea, and malabsorption.4 On biopsy, the gastrointestinal mucosa contains excess intraepithelial lymphocytes, villous blunting, lymphoid aggregates, granulomas, crypt distortion, and, as noted above, a characteristic lack of plasma cells.12,13 Another common feature is villous flattening in the small intestine, suggesting celiac sprue, but in our experience, wheat exclusion is not likely to be helpful. Nodular lymphoid hyperplasia (containing an expanded number of B cells but reduced plasma cells) is common and can impair the absorption of nutrients. Nutrient replacement, broad-spectrum antibiotics, or, in some cases, oral budesonide can be of use in refractory cases.
Lymphoma and other malignancies
The overall incidence of malignancies in CVID appears to have increased, but the data for cancers other than lymphoma are difficult to separate out. In one study of 416 CVID subjects in Australia, there was a 5-fold increase in cancer overall, but lymphoma/leukemia was 31.5% of the total.17 The incidence of stomach cancer may be increased as well, but recent comparative data appear to show a reduced incidence over prior studies.6 In the US report, 8.2% of CVID patients had a lymphoid malignancy, all B cell in type. As noted in earlier reports from our group,4,6,18 lymphoma was more common in females than males (P = .04). Of these, non-Hodgkin B-cell lymphomas were the most common, with some of these being further classified into specific B-cell phenotypes, including mucosa-associated lymphoid tissue lymphoma, marginal zone lymphoma, and T cell–rich B-cell EBV-associated lymphoma. Monoclonal B lymphocytosis was noted in one patient. Lymphomas in CVID are usually extranodal, B cell in type, and, unlike lymphomas in other congenital immune defects, are more common in subjects in the fourth to seventh decade of life and are usually EBV negative4,19,20 Cancers of other sorts also developed in 33 patients (7%) in this study.6 Other large series have not noted this high a percentage of lymphoma: 1.6% in Italy,5 3% in the ESID study,2 3.8% in a previous United Kingdom study, and 2% in a combined Danish/Swedish cohort.21
Phenotypes and survival
Of the 411 subjects with known follow-up, 93 patients (19.6%) in our US cohort followed over 4 decades had died. This is clearly reduced from the 24% noted previously4 and significantly improved from the data collected by Healy et al in 1971 before the introduction of IV Ig replacement, in which there was a 29% mortality. For the ESID CVID cohort, there was a 15% mortality over a similarly extended period.2 Although the survival of subjects with CVID has improved markedly over time, it does not compare to age-matched subjects of the same sex. For the US study, 58% of female patients with CVID survived more than 4 decades, compared with 80% of age-matched female subjects; 53% of males survived compared with 68% of age-matched male subjects.6 Due to the heterogeneity of the population followed over this period, the median age at death was 44 years (range, 10-90) for female subjects and 42 years (range, 9-79) for male subjects: not significantly different. The predominant causes of death included respiratory failure from chronic lung disease, lymphoid or other malignancies, or overwhelming infections. However, as also seen in the ESID study,2 in the US group, the risk of death in follow-up was nearly 11 times higher for CVID patients with 1 or more of the noninfectious complications outlined above than for subjects who had infections only (hazard ratio [HR] = 10.96; P < .0001).6 In fact, 89 of the 93 subjects who died had one or more of the noninfectious complications and only the 4 others had infections only. Kaplan-Meier analyses confirmed this observation, with a long-term survival of 95% for patients without versus 42% for those with noninfectious complications. Not all complications appeared to be associated with reduced survival. Patients with gastrointestinal disease (HR = 2.78; P = .0004), liver disease/hepatitis (HR = 2.48; P = .0003), lymphoma (HR = 2.44; P = .001), chronic lung disease leading to functional lung disease (HR = 2.06; P = .001), or malabsorption (HR = 2.06; P = .022) had reduced survival in this interval compared with CVID patients without these particular complications. In contrast, patients with any of the autoimmune conditions, cancers other than lymphoma, history of splenectomy, presence of granulomatous disease, or the development of bronchiectasis alone, did not have significantly reduced survival over the 4 decades of study. Kaplan-Meier survival curves also affirmed these observations, showing significantly reduced survival for patients with gastrointestinal disease (P = .005), malabsorption (P = .0196), chronic lung disease (P = .0002), liver disease/hepatitis (P < .0001), and lymphoma (P < .0001), but not for subjects with a history of autoimmunity (P = .1), cancer other than lymphomas (P = .21), previous splenectomy (P = .08), the presence of granuloma in any organ (P = .78), or the radiologic observation of bronchiectasis without impaired lung function (P = .54).6
In the US study, the age at death was significantly correlated with both earlier age at symptom onset (r = 0.8; P < .0001) and younger age at diagnosis (r = 0.88; P < .0001).6 Examining immunologic parameters associated with mortality by Cox proportional hazards modeling, a lower baseline serum IgG level (HR = 0.998; P = .0079), fewer peripheral blood B cells (HR = 0.933; P = .0004), or an increased serum IgM level at diagnosis (HR = 1.005; P = .0021) were all associated with increased mortality in the 4 decades of observation.
Other laboratory studies and pathogenesis
CVID was first described more than 60 years ago, but the causes remain unclear for the majority of patients. Most subjects have normal numbers of peripheral blood B cells (but these appear immature); reduced numbers of memory B cells identified by the surface marker CD27; and, in concert with the low serum IgG and IgA, even more reduced numbers of isotype-switched memory B cells (IgD-IgM-CD27+).22,23 One the other hallmarks of CVID is a relative or complete loss of plasma cells in BM and other tissues such as the lamina propria of the gastrointestinal tract.22,24 In terms of genetics, autosomal-recessive genetic mutations in genes important for B-cell function, including inducible costimulatory (ICOS),25 CD19,26,27 B-cell activating factor (BAFF) receptor,28 CD20,29 CD21,30 and CD81,31 have been identified in a small number of patients, more commonly in circumstances of consanguinity. CD27 deficiency was also noted in 2 brothers with persistent EBV viremia and hypogammaglobulinemia in one brother, suggestive more of a combined immune defect.32 Although heterozygous and homozygous mutations in the gene for the BCR transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) have been found in 8%-10% of subjects33–35 and are significantly associated with CVID,36,37 some of the same mutations may also be found in healthy controls and non-immunodeficient relatives, suggesting that these are disease-associated polymorphisms.38 For this reason, examining this gene is not considered diagnostic of CVID or predictive of the development of immune deficiency. Recent genome-wide studies have demonstrated the unique genetic characteristics of this immune defect, showing both novel chromosomal associations and highly significant copy number loss and gain.39
Monitoring patients over time
One of the questions that have arisen is how to follow CVID subjects over time. Due to the heterogeneity of this disease, there are no set rules aside from regularly scheduled follow-ups, and periodic monitoring of serum IgG levels. Full chemistry panels and complete blood counts are also important to check for small or larger problems that can arise over time. Monitoring subjects for lung disease has been controversial and there is no current consensus, but most agree that chest X-ray at baseline is more than reasonable. However, because radiosensitivity has been demonstrated in CVID,40 MRIs appear to provide a satisfactory means of follow-up.41 For more frequent monitoring of patients with chronic cough and/or known lung damage, we do complete lung functions including carbon monoxide diffusion as a means of assessing lung damage at yearly intervals. Gastrointestinal diseases will be evident with symptoms of diarrhea and/or weight loss and this can be evaluated with appropriate measures. In our view, the data do not suggest that routine endoscopy is required, although patients with suggestive gastrointestinal symptoms should have appropriate upper and/or lower endoscopy with examination for H pylori or other mucosal changes. Specific monitoring for autoimmunity is not required because routine blood counts and general medical oversight will reveal characteristic symptoms. Lymphadenopathy is very common in CVID and evaluation of these is not simple; when new nodes appear and persist, biopsy may be required. However, lymphomas are more commonly extranodal and appear in unusual locations such as the lungs or mucosal-associated tissues such as the gastrointestinal tract, and are thus not amenable to standard follow-up measures. BM examinations to look for lymphoma have not been revealing, with the exception of advanced cases in which the diagnosis was already established by other means.
Conclusions
There have been several studies on the pathogenesis and best approaches to the treatment of CVID and its complications since it was first recognized as a syndrome more than 60 years ago. Increasing independence due to the use of home-care Ig therapy has improved the quality of life for all CVID patients. The majority of patients can expect to have stable health, attend school, have career expectations, and perform almost any job; however, periodic planned follow-up for global assessment is still required.
Acknowledgments
This work was supported by the National Institutes of Health (AI 101093, AI-467320, and AI-48693; National Institute of Allergy and Infectious Diseases contract 03-22; and the David S. Gottesman Immunology Chair).
Disclosures
Conflict-of-interest disclosure: The author has consulted for Baxter, Grifols, and Merck. Off-label drug use: None disclosed.
Correspondence
Charlotte Cunningham-Rundles, Immunology Institute and the Departments of Medicine and Pediatrics, Mount Sinai School of Medicine, 1425 Madison Ave, Rm 11-20, New York, NY 10029; Phone: 212-659-9268; Fax: 212-987-5593; e-mail: charlotte.cunningham-rundles@mssm.edu.
