
Abstract
Hemophilia A is a rare bleeding disorder treated with numerous factor VIII (FVIII)–containing replacement concentrates. This treatment approach has led to the formation of alloantibodies that neutralize the FVIII activity (inhibitors) conveyed by these commercially available concentrates in ∼ 25% of patients with severe hemophilia A (FVIII activity < 1% of normal). This phenomenon significantly complicates the treatment of these patients and compromises the effectiveness and efficiency of these products to reverse or prevent bleeding complications. Studying the population with alloantibody inhibitors is imperative but difficult due to the overall small number of individuals affected and the heterogeneity within this limited group. Furthermore, few randomized clinical trials have been conducted to answer pertinent questions so many controversies persist. This article focuses on the conflicting data on the variables associated with alloantibody FVIII inhibitor development with a particular emphasis on age and intensity of first treatment, the role of primary prophylaxis regimens in modulating this phenomenon, and the degree of purity of FVIII product as a potential contributing risk factor. The optimal dosing regimen and type of FVIII replacement product that should be used to achieve the highest success rate in immune tolerance induction (ITI) protocols are also discussed, as well as whether the addition of immunomodulatory agents, especially rituximab, to ITI regimens enhances the durability of ITI and the eradication of alloantibody FVIII inhibitors.
Introduction
Hemophilia is a rare congenital disease affecting 1 in 10 000 live male births with hemophilia A and 1 in 30 000 with hemophilia B. Numerous factor VIII (FVIII) gene mutations have been identified, resulting in different severities of disease. Whereas many mild cases (FVIII activity > 5%) may not be diagnosed until later in life after trauma or surgery or not at all, most moderate (FVIII 1%-5%) and severe (FVIII < 1%) cases are likely known at birth or early in life and are usually followed in comprehensive hemophilia care centers. Individuals with severe hemophilia, by nature of the disease, use most of the hemophilia care center resources and have been the focus of most of the research in this area. A specific area of concern has been the formation of alloantibodies against replacement FVIII concentrates, the mainstay of treating and preventing bleeding in these patients. This occurs in approximately 30% of patients with severe hemophilia A. Often these alloantibodies, also called inhibitors, render the use of such replacement ineffective and alternative treatments are costly and less effective. An intense research focus has been on factors contributing to inhibitor development in an attempt to reduce their emergence and on inhibitor eradication. The question has been raised of whether age or intensity at first FVIII concentrate exposure or at the start of early prophylactic dosing could influence the risk of inhibitor development. It is also being debated whether different commercially available FVIII products may have various immunogenicities, especially comparing plasma-derived versus recombinant concentrates. Once an inhibitor emerges, immune tolerance induction (ITI) in the form of daily doses of FVIII in an attempt to achieve immunogenic acceptance is the standard treatment. It is not successful in all patients (only 60%-80% respond) and often has to be given over many months. The best choice of product when starting ITI, the optimal dose, and whether the addition of immunomodulating agents is of benefit have not yet been determined.
Severe hemophilia A is not a homogenous disease and patients with severe hemophilia have different FVIII gene mutations, racial and ethnical backgrounds, environmental influences, and immunologic make-up. Only a few randomized controlled trials have been conducted in severe hemophilia. Due to the rarity of well-characterized hemophilic populations (predominantly in economically developed nations), accrual to clinical studies is typically challenged and frequently the cohort sizes do not meet traditional biostatistical requisites. Alternative biostatistical approaches, such as Bayesian techniques rather than confidence interval methods, may be more efficient and practical in hemophilia clinical research. A well-conducted study on the effect of FVIII prophylaxis on joint health, for example, took almost 4 years to recruit 65 patients with severe and moderately severe hemophilia A and took 9 years to complete.1 To recruit enough subjects to a randomized controlled trial often necessitates national and international collaborations, which themselves face increasingly strict ethical and regulatory constraints. Many countries and local ethical boards vary widely in their requirements and there is no standardization. Furthermore, many recommendations concerning hemophilia treatment are not evidence based, but rather are devised from experimental and often anecdotal case reports, small single-center studies, retrospective reviews, and registries. Major controversies in the field therefore remain to be studied and resolved.
Inhibitor development
It has been shown repeatedly that disease severity, major FVIII gene defects, family history, and non-Caucasian race are major risk factors for inhibitor development.2,3 Inhibitors tend to develop within the first 10 exposure days (defined as a 24-hour period during which one or more exposures to FVIII therapy was recorded) and rarely develop after 50 exposure days. Beyond that, there are several controversial points that are thought to favor antibody production, as discussed below.
Age at first treatment
Early FVIII exposure has been implicated as a possible contributor to alloantibody FVIII inhibitor development. This may be of particular concern in the United States, where many infants are considered for early surgical intervention (circumcision or port placement for prophylaxis) for which factor concentrate is needed.
The potential impact of age at first exposure on inhibitor development was assessed in 3 retrospective cohort studies summarized in Table 1. In a Spanish cohort of 62 children with severe (FVIII < 2 IU/dL) hemophilia A, 24% of whom developed persistent inhibitors, a statistically significant difference (P = .03) in the cumulative incidence of laboratory-detected inhibitor formation based on age at first exposure was demonstrated.4
Summary of 3 retrospective cohort studies on the potential impact of age at first exposure on inhibitor development in severe hemophilia A
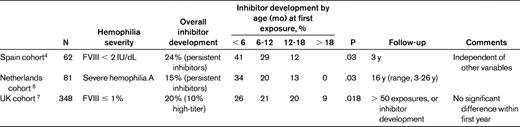
A cohort study from the Netherlands of 81 infants with severe hemophilia A confirmed this trend, assessing the incidence at 100 exposure days.5 This trend was again demonstrated in a larger multicenter review of 366 patients with severe hemophilia A in the Netherlands (the CANAL cohort study), but disappeared when adjusting for confounding factors, especially intensity at first treatment.6 This study selected for symptomatic patients (decreased FVIII half-life) but did not account for a large heterogeneity in FVIII products used.
In another retrospective cohort study from the United Kingdom of 348 children with severe hemophilia A (FVIII ≤ 1%), 20% developed an inhibitor and 10% of these were high titer. When correlating this to age at first exposure, there was a significant decrease in inhibitor development when children were treated after they were 18 months old (20%-26% at ages 0-18 months, 9% at age > 18 months). In that study, however, exposure to FVIII during the neonatal period was not associated with a higher incidence of inhibitors than treatment later during the first year of life.7
An Italian case-control study of children treated exclusively with recombinant FVIII compared 60 children with inhibitors to 48 inhibitor-free controls. Although there seemed to be an increased trend for inhibitor formation if patients were treated at an early age, this did not hold true after adjustment for genetic factors.2
In summary, there is some concern over factor exposure in the first year of life contributing to alloantibody formation. However, the data are conflicting and some studies conclude that there are confounders such as more severe genotype, phenotype, or intensity of treatment that explain the increased risk for inhibitor development rather than the timing of the exposure. An emerging concept is that “danger signals” contribute to inhibitor risk. This can happen when antigen presenting cells, which are necessary for the initiation of immune responses, are induced by “danger signals ” such as infection, vaccinations, tissues undergoing stress, or damage.8
Intensity of early treatment
After assessing whether age at first treatment is a deciding factor in inhibitor development, the question arose of whether treatment intensity during early treatment could also be a predictor for inhibitor formation.
The CANAL study reported a 3.3-fold higher risk for inhibitor development in patients treated for at least 5 consecutive days at first FVIII exposure over those receiving 1 to 2 days of treatment. This was particularly true at higher (> 50 IU/kg) doses and when the first treatment was for surgery rather than for bleeds or prophylaxis.6
The above-mentioned retrospective case control study in the United Kingdom examined early treatment patterns in severe hemophilia A (FVIII ≤ 1%) in 78 (63 high-titer) inhibitor patients and age-matched controls and found that high frequency treatment (FVIII on > 50% of days) increased inhibitor risk by approximately 2.5-fold. Similarly, high treatment intensity per episode resulted in increased inhibitor risk: the overall risk for more than 5 exposure days per episode was 2.7 and for more than 10 exposure days, it was 5.5. This was true after treatment for life-threatening conditions but not for surgeries such as Port-A-Cath insertion.3
Prophylaxis
As FVIII prophylaxis is increasingly available and considering the potential danger signals of early intense treatment, the question of whether primary prophylaxis, and thus early, controlled antigen presentation, could prevent inhibitor formation is pertinent.
The Italian multicenter experience of hemophilia A (FVIII 1%-2%) patients showed a significantly smaller risk for inhibitor development in those treated with prophylaxis over on-demand treatment,2 a trend that was also confirmed in the CANAL study6 but not in the UK study.3
In a prospective study of hemophilia A (FVIII ≤ 2%), boys < 30 months of age were randomized to receive 25 IU/kg of FVIII every other day versus on-demand treatment. In the prophylaxis arm, 2 of 32 developed an inhibitor, whereas none (0 of 33) did in the on-demand arm.1
A prospective pilot study in Germany compared low-dose prophylaxis of 25 U/kg weekly at the first sign of bleeding to more standard prophylaxis of 40-50 U/kg 3 times a week starting with the first joint or severe bleed. The groups had no significant differences in ethnicity, mutation type, product type used, age at or reason for first exposure, surgeries within the first 20 exposure days, or timing of vaccinations. Only 1 of 26 patients in the low-prophylaxis group developed inhibitors, compared with 14 of 30 in the control group. None of the patients in the low-prophylaxis group had high-titer inhibitors, but 8 of 30 in the control group did.9
Product choice in the treatment of severe hemophilia A
Transmissions of blood-borne pathogens and inhibitor formation have been the most detrimental complications associated with the use of plasma-derived FVIII concentrates since their commercial introduction. Although the infectious disease risk has been minimized through various regulatory screening practices of potential donors to the source or recovered plasma pool, the inclusion of effective viral inactivation/elimination techniques in the purification and manufacturing processes for plasma-derived FVIII concentrates, and ultimately by the advent of genetically engineered recombinant products, the inhibitor risk persists.
The use of FVIII concentrates, especially recombinant products, is on the rise (National Blood Authority Annual Report 2007-2008), making up more than 90% of the market share in the United States. Improved safety of plasma-derived products has been achieved over the last decade through more rigorous donor screening, plasma testing, immunoaffinity, pasteurization, use of solvent/detergents, and nanofiltration. Despite this improving safety profile, the risk for newly emerging and reemerging infections is not negligible,10 and is still a major concern for patients and parents when choosing a FVIII product, outweighing the concern for developing an inhibitor.11
Emerging preclinical studies and several cases, cohorts, and retrospective reviews raise some concern about an increased incidence of inhibitor formation with the use of recombinant products, but no prospective trials investigating this have been conducted. This is largely because there was a significant shift to recombinant FVIII (rFVIII) products in most developing countries after the devastating experience with infections due to plasma-derived FVIII (pdFVIII). To compare the 2 classes of products in a randomized trial was not thought to be ethical for many years. It was not until the emergence of data suggesting increased immunogenicity with rFVIII that pdFVIII was again considered to be an equal treatment option.
Several basic immunologic principles have been explored that give scientific merit to the hypothesis that pdFVIII, especially VWF-containing FVIII products, may be less immunogenic. These principles include antigenic competition, epitope masking, protection from endocytosis, and variations in the cytokine environment; these are summarized in Table 2 and outlined below.
Summary of basic immunologic principles giving scientific merit to the hypothesis that pdFVIII products may be less immunogenic than rFVIII products

The theory of antigenic competition is based on the fact that plasma-derived products contain other proteins in addition to FVIII that may distract an immune response away from the FVIII molecule. Alloantibody binding has been shown to target certain regions (epitopes) of the FVIII molecule, specifically the A2, A3, and C2 domains. Epitope masking conceivably happens when VWF attached to FVIII masks the C2 domain on the FVIII molecule, an area identified to be significant in antibody production. This was demonstrated when inhibitor titers were found to be considerably higher in response to an rFVIII concentrate than to a pdFVIII/VWF concentrate in patients with FVIII C2 domain specificity.12 This theory, however, does not account for alloantibodies directed to the A2 or A3 domain. It has also been shown that the addition of VWF to FVIII appears to protect against endocytosis of FVIII by dendritic cells (APCs), and thus prevents presentation to CD4+ cells and subsequent antibody production.13
An elegant series of experiments was conducted in hemophilic mice, in which both product types caused an up-regulation of gene expression of proinflammatory cytokines. However, mice receiving rFVIII had 2-fold higher anti-FVIII inhibitor levels than those treated with pdFVIII/VWF, and overall displayed a different cytokine profile, likely triggering a different immune response. Whereas rFVIII produced higher levels of IL-2, IL-10, and IFN-γ, pdFVIII resulted in higher levels of IL-4, IL-5, and TGF-γ.14
There are many inherited and environmental factors contributing to inhibitor development. Two systematic reviews published in 2003 showed a significantly lower incidence of inhibitor development with the use of single pdFVIII product over a single rFVIII product; these reviews are summarized in Table 3.15,16 However, after adjusting for study design, study period, testing frequency, and median follow-up, these data lost statistical significance in the second larger review.16
Summary of 2 systematic reviews published in 2003 showing a significantly lower incidence of inhibitor development with the use of single pdFVIII product over a single rFVIII product

n/a indicates not available.
The above data are largely retrospective, and a multivariate analysis of the studies in the second review showed an overestimation of inhibitor development in the retrospective studies. All studies encompass different modalities of inhibitor testing, and there is large heterogeneity in study populations with various disease severities, mutations, and other inhibitor risk factors such as ethnicity, age at first exposure, etc. They also comprise a variety of plasma-derived products, involving various purities and the presence and absence of VWF. Length of follow-up in the studies varied and there were additional confounding factors.
Franchini et al published a systematic review and meta-analysis on published prospective studies earlier this year. This included data from 25 prospective studies and 800 patients and found that the inhibitor rate between previously untreated patients with severe hemophilia A did not vary significantly (21% vs 27% of overall inhibitors and 14% vs 16% of high-titer inhibitors).17
Whereas the immunogenicity of pdFVIII versus rFVIII products is being examined in a multicenter clinical trial (see discussion of the SIPPET study in the conclusion), a major evolving controversy is over inhibitor development based on constructs of the rFVIII concentrates and mismatches to the host's FVIII genetic polymorphism. It was shown recently that there are 6 wild-type FVIII proteins (haplotypes H1-H6), which are differentiated only by single-nucleotide polymorphisms. It appears that Caucasians exclusively have wild-type H1 and H2, where other races also express the other haplotypes. The pdFVIII mainly contains haplotype H1 and H2 (likely due to the predominantly Caucasian donor pool), and rFVIII is of either the H1 or the H2 haplotype. This could explain the significantly higher inhibitor occurrence in the African-American population, and is currently being investigated in detail.18
Inhibitor treatment
Based on registry data, there is general consensus that peak inhibitor titer, inhibitor titer at the start of ITI, and time from inhibitor onset to initiation of ITI can all influence the success of immune tolerance. Controversy remains as to whether the FVIII dose and dosing regimen, choice of FVIII product, and use of adjunctive therapies contribute to successful ITI. Details on these debates were published previously19 and are ongoing.
Factor VIII dosing and regimen
Investigators have reported comparable rates of successful tolerization ranging from 63%-91% despite a wide variety of ITI regimens.20–24 Two large registries reported conflicting data when examining ITI success in relation to FVIII dosing. The International Immune Tolerance Registry reported a statistically improved outcome with FVIII dosing of 200 units/kg/d, but the North American Immune Tolerance Registry reported better ITI success with a lower ITI regimen.25,26 To answer this question prospectively, the International Immune Tolerance Induction (I-ITI) study was launched in 2002. This multicenter study set out to randomize 150 good-risk patients with severe hemophilia A to receive either high-dose (200 IU/kg/d) or low-dose (50 IU/ kg/thrice weekly) ITI.27 The study closed enrollment in 2010 because more bleeding was noticed in the low-dose ITI arm and because of concerns over the accrual needed to reach the statistical end points.
Product choice
The scientific debate around product choice for ITI follows the same underlying mechanistic logic for product choice for first treatment described above.
In a German study, 8 of 10 patients who had unsatisfactory responses to ITI with monoclonal or recombinant FVIII subsequently had complete inhibitor eradication and tolerance with the use of a VWF/FVIII concentrate. Similar findings were reported in 2 other German centers, where overall, 23 of 28 patients (82%) undergoing ITI with VWF/FVIII concentrate were successfully tolerized, compared with only 6 of 14 patients (43%) treated with monoclonal or recombinant FVIII.28
In an Italian study of 17 patients, 9 (53%) with poor prognostic features for response were successfully tolerized using a VWF/FVIII concentrate. Another 7 patients achieved partial success with sustained low inhibitor titers but abnormal FVIII recovery or half-life.29
These reports reviewed small numbers of subjects, and the claim that VWF-containing FVIII products might lead to improved ITI remains controversial. A recent critical review of the literature could not support that claim.30
An international, prospective, controlled, randomized, open-label study is currently under way to compare FVIII/VWF concentrates with FVIII concentrates at 200 IU/kg/d on their ability to induce immune tolerance in patients with hemophilia A with high responding inhibitors and poor prognosis for success who have not previously undergone ITI (RESIST naive study, registered at www.ClinicalTrials.gov as NCT01051544). For patients who have failed previous ITI the RESIST-experienced trial (NCT01051076) is trying to determine whether VWF-containing concentrates can achieve successful ITI in this patient population.31
Immunomodulation
Whereas 50%-80% of patients with inhibitors respond to traditional ITI, there remains a significant number who do not. The addition of immunomodulatory measures has been considered for years. A routinely used ITI protocol in Sweden (the Malmö protocol) combines FVIII administration with immunosuppression. Extracorporeal immunoadsorption is used as needed to remove high-titer inhibitors and to achieve an inhibitor titer below 10 BU at the initiation of ITI. Most patients are then given a single dose of oral corticosteroids (50-150 mg), followed by cyclophosphamide (12-15 mg/kg IV for 2 days, then orally 2-3 mg/kg/d for an additional 8-10 days) and IV immunoglobulin (0.4 mg/kg/d for 5 days) with the start of high daily doses of FVIII.21 Unanswered is whether the addition of such agents can reduce the time and intensity of ITI needed and whether this would aid in tolerizing individuals who did not succeed with traditional ITI with FVIII alone. There are emerging concerns that one ITI regimen does not fit all; that is, inhibitor immunology is complicated and involves many different pathways.32 It therefore behooves us to gain a better understanding of these pathways and mechanisms to tailor the ITI regimen to each individual patient.
An immunomodulatory agent that is of particular interest is rituximab, a chimeric monoclonal antibody against the CD20 antigen that blocks the proliferation of normal B cells, thus interfering with IgG antibody production. Several case reports on the use of rituximab as part of an attempt at inhibitor eradication, either alone or in combination with ITI, have been published. A summary and analysis of 29 studies, including 49 case reports, showed that a durable complete remission was obtained in 53% of cases and no severe adverse events related to rituximab were recorded.33
A national survey of all 23 comprehensive care hemophilia centers in the United Kingdom reported 15 consecutive severe (< 1% FVIII) hemophilia A patients with loss of a detectable inhibitor in 40% of patients on rituximab in conjunction with ITI, but a lasting response was seen in only 2 patients.34
In the United States, a prospective, multicenter, National Institutes of Health (NIH)–funded study investigating the efficacy of rituximab as a single agent in patients with severe hemophilia A and high responding inhibitors who previously failed ITI was closed by the NIH data and safety monitoring board because it was felt that it would not enroll enough subjects to have the desired statistical power. The above-mentioned review from the United Kingdom suggested that rituximab may be more successful if it was used in conjunction with ITI.34 Currently, a registry through the Hemophilia and Thrombosis Research Society (https://www.htrsregistry.org/login.html) is collecting data on congenital hemophilia patients who have been treated with rituximab to gain further insights on how best to use rituximab in this population (with or without ITI), what predictors for success might be, and to elicit possible side effects (R.K.-J., principal investigator).
Whereas the response rate to rituximab adjunctive therapy is intriguing, data interpretation is complicated by the variable FVIII regimens used, different definitions of response, various durations of follow-up, and the potential for preferential reporting. Furthermore, the long-term adverse effects of B-cell depletion, particularly in children, are unknown. Prospective data from patients treated with a standardized protocol are needed to clarify the role of rituximab in ITI.
Conclusion/Discussion
The most studied cohort in bleeding disorders is that of severe hemophilia A. Although geographically, racially, and ethnically relatively evenly distributed, severe hemophilia A is rare and in itself quite heterogeneous. To conduct large randomized controlled trials is difficult, so our current treatment recommendations are often based on smaller cohort studies, retrospective reviews, case reports, and registries. Many of the reports are contradictory, leaving plenty of room for controversy. This review highlights some of these controversies with particular attention to inhibitor development and eradication.
Several multicenter, retrospective reports have assessed the role of age at first exposure to FVIII, and all showed a trend toward a higher rate of inhibitor development if treatment with FVIII occurred at a younger age, especially < 6 months of age. However, one study did not find an increased risk for inhibitors with treatment in the neonatal period, another could not verify this trend when adjusting for genetic mutations, and a third failed to demonstrate the trend after adjusting for other confounding factors.
Treatment intensity and FVIII dose may contribute to inhibitor development. Surgery appears to be a high-risk situation, but this does not seem to include Port-A-Cath placement. These data are based on relatively small, retrospective cohorts and need to be verified.
It is not clear whether early prophylaxis lessens the risk of inhibitor development, as some retrospective data and a prospective pilot study have suggested.
Over the last decade, the use of FVIII has increased, especially the use of rFVIII. The important clinical question of whether pdFVIII products result in fewer inhibitors over rFVIII products remains. To answer this question, a multicenter, randomized, open label, clinical trial in previously untreated patients or minimally blood component–treated patients with severe hemophilia A (FVIII < 1%) is ongoing. The trial is investigating the rate of inhibitor formation in the first 50 exposure days of VWF/FVIII compared with rFVIII concentrates (“Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET)”; www.ClinicalTrials.gov identifier NCT01064284).
Once inhibitors develop, immunotolerance with repeated dosing of FVIII has become a standard therapy; however, no standard dosing schedule has been defined. Conflicting data from registries favor very high versus very low dosing schedules. After similar scientific arguments as those regarding the choice of factor product in initial treatment, it is proposed that VWF-containing FVIII products may have better success in ITI, but prospective trials to verify this are ongoing. Considering the heterogeneity of severe hemophilia A patients with inhibitors and the complexity of each individual immune system, the addition of immunomodulating agents to ITI regimens adds a new tier of questions to those not yet definitively answered. While we continue to conduct clinical trials that are often difficult to accomplish and may or may not reach their intended end point, we would be well served to collect ancillary immunologic data to better understand the complexity of these issues from diverse perspectives.
Disclosures
Conflict-of-interest disclosure: The author has consulted for and received honoraria from Novo Nordisk, Inspiration, Grifols, and Baxter, and is a member of the Globally Emerging Hemophilia Expert Panel, which is sponsored by Bayer. Off-label drug use: Rituximab for inhibitor treatment.
Correspondence
Rebecca Kruse-Jarres, MD, MPH, Section of Hematology and Medical Oncology, Tulane University School of Medicine, 1430 Tulane Ave, SL78, New Orleans, LA 70112; Phone: (504) 988-3562; Fax: (504) 988-5483; e-mail: rkruseja@tulane.edu.
