
Abstract
Heparin-induced thrombocytopenia (HIT) is a prothrombotic drug reaction caused by platelet-activating IgG antibodies that recognize platelet factor 4 (PF4)/polyanion complexes. Platelet activation assays, such as the serotonin-release assay, are superior to PF4-dependent immunoassays in discerning which heparin-induced antibodies are clinically relevant. When HIT is strongly suspected, standard practice includes substituting heparin with an alternative anticoagulant; the 2 US-approved agents are the direct thrombin inhibitors (DTIs) lepirudin and argatroban, which are “niche” agents used only to manage HIT. However, only ∼ 10% of patients who undergo serological investigation for HIT actually have this diagnosis. Indeed, depending on the clinical setting, only 10%-50% of patients with positive PF4-dependent immunoassays have platelet-activating antibodies. Therefore, overdiagnosis of HIT can be minimized by insisting that a positive platelet activation assay be required for definitive diagnosis of HIT. For these reasons, a management strategy that considers the real possibility of non-HIT thrombocytopenia is warranted. One approach that I suggest is to administer an indirect, antithrombin (AT)–dependent factor Xa inhibitor (danaparoid or fondaparinux) based upon the following rationale: (1) effectiveness in treating and preventing HIT-associated thrombosis; (2) effectiveness in treating and preventing thrombosis in diverse non-HIT situations; (3) both prophylactic- and therapeutic-dose protocols exist, permitting dosing appropriate for the clinical situation; (4) body weight–adjusted dosing protocols and availability of specific anti-factor Xa monitoring reduce risk of under- or overdosing (as can occur with partial thromboplastin time [PTT]–adjusted DTI therapy); (5) their long half-lives reduce risk of rebound hypercoagulability; (6) easy coumarin overlap; and (7) relatively low cost.
Introduction
Hematologists are often asked to help diagnose and treat heparin-induced thrombocytopenia (HIT). This scenario can be challenging: although thrombocytopenia is common in hospitalized patients, HIT represents only a small subset. Furthermore, the most widely used tests to detect HIT antibodies, platelet factor 4 (PF4)–dependent enzyme-immunoassays (EIAs), frequently detect clinically irrelevant antibodies, with potential for overdiagnosis. In addition, the U.S. Food and Drug Administration (FDA)–approved anticoagulants for treatment of HIT, direct thrombin inhibitors (DTIs), are rarely used outside of the HIT arena. This presents a dilemma when only ∼ 10% of patients who are investigated serologically for HIT actually have this diagnosis. Moreover, partial thromboplastin time (PTT) monitoring of DTI therapy can be confounded by coagulopathy either due to comorbidity (eg, liver dysfunction) or even severe HIT itself (“HIT-associated consumptive coagulopathy”); in such patients, DTI therapy can be ineffective due to systematic underdosing.
In this chapter, I describe my approach for the diagnosis and management of suspected HIT focusing on 2 issues: (1) the use of a washed platelet activation assay such as the serotonin-release assay (SRA) to ascertain which patient really has HIT, and (2) the use of an antithrombin (AT)–dependent factor Xa inhibitor (danaparoid or fondaparinux), a treatment approach that avoids certain pitfalls of DTI therapy but, more importantly, offers the known safety and benefit of these agents for the large majority of patients who turn out not to have HIT (note: fondaparinux is an “off-label” therapy for HIT).
Central paradigm of HIT
The HIT central paradigm in 20111 : HIT is caused by antibodies of the IgG class that produce strong activation of platelets via the platelet FcγIIa (IgG) receptors; these antibodies recognize large multimolecular complexes of PF4 bound to heparin (PF4/H) and are already detectable in patient serum/plasma at the beginning of the HIT-associated platelet count decline.2 Almost always, patients have undergone a proximate immunizing exposure to unfractionated or (less commonly) low-molecular-weight heparin.
Platelet-activating antibodies comprise a subset of anti-PF4/H antibodies
Only a minority of anti-PF4/H antibodies are platelet-activating. Here, the “iceberg model” of immunization is apt (Figure 1): just as only 1/10th of an iceberg protrudes above the waterline, so too do only ∼ 10% of anti-PF4/H antibodies identified in certain patient cohorts have platelet-activating properties (discussed subsequently).3,4 Washed platelet assays are superior to conventional platelet aggregometry (using citrated platelet-rich plasma) for detecting HIT antibodies: in North America, the SRA is the most common washed platelet assay, whereas in central Europe (eg, Germany and Austria), a platelet aggregation end point is more often used (ie, the heparin-induced platelet activation [HIPA] test).

“Iceberg model” of HIT. Clinical HIT, comprising HIT with (HIT-T) or without thrombosis, is represented by the portion of the iceberg above the waterline; the portion below the waterline represents subclinical anti-PF4/heparin seroconversion. Three types of assays are highly sensitive for the diagnosis of HIT: the washed platelet activation assays (SRA and HIPA), the IgG-specific PF4-dependent EIAs (EIA-IgG), and the polyspecific EIAs that detects anti-PF4/heparin antibodies of the 3 major immunoglobulin classes (EIA-IgG/A/M). In contrast, diagnostic specificity varies greatly among these assays, being the highest for the platelet activation assays (SRA and HIPA) and lowest for the EIA-IgG/A/M. This is because the EIA-IgG/A/M is most likely to detect clinically irrelevant, non-platelet-activating anti-PF4/heparin antibodies. The approximate probability of SRA+ status in relation to a given EIA result, expressed in OD units, was obtained from the literature.7
“Iceberg model” of HIT. Clinical HIT, comprising HIT with (HIT-T) or without thrombosis, is represented by the portion of the iceberg above the waterline; the portion below the waterline represents subclinical anti-PF4/heparin seroconversion. Three types of assays are highly sensitive for the diagnosis of HIT: the washed platelet activation assays (SRA and HIPA), the IgG-specific PF4-dependent EIAs (EIA-IgG), and the polyspecific EIAs that detects anti-PF4/heparin antibodies of the 3 major immunoglobulin classes (EIA-IgG/A/M). In contrast, diagnostic specificity varies greatly among these assays, being the highest for the platelet activation assays (SRA and HIPA) and lowest for the EIA-IgG/A/M. This is because the EIA-IgG/A/M is most likely to detect clinically irrelevant, non-platelet-activating anti-PF4/heparin antibodies. The approximate probability of SRA+ status in relation to a given EIA result, expressed in OD units, was obtained from the literature.7
Platelet-activating antibodies are IgG and usually present at high levels
Only IgG antibodies can activate platelets, which is why IgG-specific commercial EIAs are now marketed.5,6 Increasing optical density (OD) values, a marker of antibody levels, correspond to a greater risk of HIT: for every 1.0 U increase in OD, the probability of a positive SRA increases by 40 (odds ratio), or, expressed another way, from < 5% to ∼ 25% to ∼ 95% for positive OD values of 0.5, 1.5, and 2.5, respectively.7 This strong relationship between OD values and a positive platelet activation assay can be seen with different EIAs and different washed platelet activation assays.8 Although IgG-specific assays have greater diagnostic specificity than do polyspecific EIAs (detecting IgG, IgA, and IgM), a strong positive polyspecific EIA is far more predictive of HIT than is a weakly positive IgG-specific assay.9
Detectability of HIT antibodies precedes thrombocytopenia
Studies of serial blood samples reveal that antibodies are readily detectable at the onset of the HIT-associated platelet count decline.2 Moreover, antibodies of different immunoglobulin classes become detectable simultaneously: that is, there is no IgM precedence.2,10 Compared with other immunohematologic conditions such as (auto)immune thrombocytopenia, HIT serology is highly sensitive. This has implications for choosing when (and when not) to test for HIT and how to interpret test results.
HIT diagnostic considerations
HIT is a “clinical-pathological syndrome,” which means that the diagnosis requires a compatible clinical picture plus detectability of platelet-activating antibodies.1 In most centers, however, a platelet activation test is not routinely performed and the EIA is used as a surrogate. This practice contributes to HIT “overdiagnosis.”
HIT overdiagnosis
Sometimes a clinician will diagnose HIT even when sensitive assays are negative (insisting that HIT is a “clinical diagnosis”) or will assume that any patient with a positive EIA must have HIT without taking into account the strength of the EIA result. Other factors contributing to HIT overdiagnosis include: (1) ordering tests in low pretest probability situations; (2) ordering automatic repeat HIT antibody testing; and, most importantly; and (3) not insisting on a positive platelet activation assay for diagnosis.
Assessment of pretest probability
Clinical scoring systems have been developed to assess the pretest probability of HIT. One example is the 4Ts scoring system,11 based on the mnemonic, Thrombocytopenia, Timing, Thrombosis (or other sequelae of HIT such as anaphylactoid reaction), and (lack of) oTher explanations. A more recent system is the HIT Expert Probability (HEP) score.12 The scores are especially useful at ruling out a diagnosis of HIT, because a low score indicates a < 2% probability of having a positive platelet activation assay (indeed, testing for anti-PF4/heparin antibodies may not be warranted in this situation). Even when the probability of HIT is deemed “high,” only ∼ 50% of patients have a positive confirmatory platelet activation assay.9,12
Low pretest probability of HIT is a feature of most intensive care unit (ICU) patients; in this setting, thrombocytopenia usually occurs too early to be HIT and there is usually at least one compelling non-HIT explanation for thrombocytopenia. Overall, the probability of HIT being the explanation for thrombocytopenia in the ICU is only approximately 1 in 100.13 However, some studies of DTI therapy for suspected HIT indicate that the majority (75%-80%) of those treated were ICU patients,14,15 inferring overdiagnosis.
Automatic repeat testing
Sometimes, routine repeat EIA testing is done a few days after an initial negative test result. This is problematic: the initial negative EIA essentially rules out HIT (high negative predictive value); routine repeat testing converts the situation into a “serosurveillance” study: therefore, a subsequent positive test usually means clinically irrelevant seroconversion.
To illustrate, consider a study by Selleng et al of postcardiac surgery patients with early-onset and persisting thrombocytopenia.16 These investigators showed that subsequent positive tests did not indicate a diagnosis of HIT; in these patients, the frequency of seroconversion matched that of controls exhibiting the expected pattern of early transient postoperative thrombocytopenia. When the platelet count remains low in a patient with a complicated postoperative course, it is usually for non-HIT reasons, and the development of antibodies therefore usually reflects subclinical seroconversion. Of course, if a subsequent new platelet count decrease occurs within the characteristic day 5-10 HIT “window” that is superimposed on the profile of early-onset and persisting thrombocytopenia, this can indicate a diagnosis of “true HIT.”17
SRA superior to EIA
The SRA, a washed platelet activation assay developed > 25 years ago at McMaster University,18 was applied in a blinded fashion to serial blood samples obtained in a large clinical trial of heparin thromboprophylaxis (hip replacement surgery).19 In this study (performed before the development of the EIA), a positive SRA strongly predicted for thrombocytopenia that began 5 or more days after starting heparin.
Subsequently, when the EIA was developed, it was applied to the archived blood samples from this trial.20 Table 1A compares the frequency of thrombocytopenia among EIA+ patients in this trial in relation to SRA status. EIA+ status alone (ie, without an SRA+ result) did not predict for thrombocytopenia. Although the IgG-specific EIA detected fewer irrelevant antibodies, it nevertheless detected more irrelevant antibodies than did the SRA.
Frequency of thrombocytopenia (>50% platelet count decrease) among EIA+ patients (polyspecific or IgG-specific assay) who received heparin (unfractionated or low-molecular-weight): a comparison of SRA+ versus SRA− status
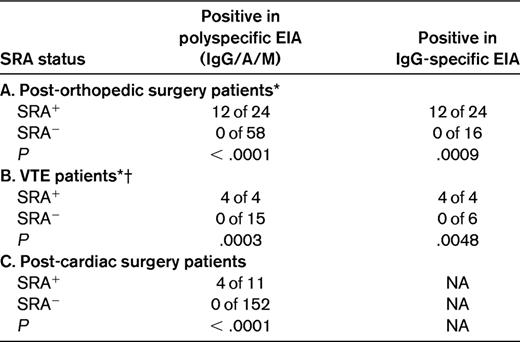
The data are consistent with the SRA having a high sensitivity for HIT (> 95%); the specificity of the SRA depends on the clinical situation, but in most circumstances is at least 90%. Patients in studies “A” and “B” were tested in both the polyspecific and IgG-specific assays. Data used to construct this table were obtained from the published literature.3,19–21
NA indicates not available.
*For the data shown, the cutoff for a positive SRA was 20% serotonin release. For study A (post-orthopedic surgery), if instead a 50% serotonin-release cutoff is used, the comparisons (polyspecific EIA) yield similar results: 11 of 20 versus 1 of 62 (P < .0001), and unchanged data for study B (VTE patients).
†For the VTE study, EIA results were ≥ 1.0 units of OD.
EIA+/SRA− status is inconsistent with clinical HIT
Clearly, SRA+ status confers risk of thrombocytopenia attributable to HIT, but what about a thrombocytopenic patient investigated for HIT who tests EIA+/SRA−? How confident can the clinician be that this profile indicates a non-HIT disorder as the explanation for thrombocytopenia?
We approached this issue by comparing the outcomes of 3 patient groups identified by investigating 100 consecutive patients referred to a hematology service because of clinically suspected HIT: EIA+/SRA+ (n = 16), EIA+/SRA− (n = 16), and EIA−/SRA− (n = 68).22 We found that only EIA+/SRA+ patients had a high frequency of thrombosis (69%). In contrast, the other 2 SRA− groups had much lower thrombosis rates (∼ 7% each), but relatively high mortality rates (∼ 25% each vs only 6% for the EIA+/SRA+ patients). These data infer that EIA+/SRA− status does not indicate HIT, but rather the presence of a non-HIT thrombocytopenic disorder (including high mortality characteristic of severely ill, non-HIT patients23 ).
Integrating clinical and laboratory information
Ideally, a diagnosis of HIT should only be made if the clinical picture is reasonably consistent with this diagnosis (generally, ≥ 4 points in the 4Ts scoring system), with the presence of platelet-activating anti-PF4/H antibodies (EIA+/SRA+ status). Moreover, there should be no non-HIT disorder judged to be a better explanation for the patient's overall clinical course.
If only an EIA test result is available, a negative test should be viewed as ruling out HIT; routine repeat testing should not be performed. An OD level of > 2.00 units indicates a high probability (> 90%) of a positive platelet activation assay and, most likely, true HIT (Figure 1). For patients with weak (0.45-0.99) or moderate (1.00-1.99) OD levels, referral of patient serum for a washed platelet activation test should be strongly considered.
Few patients with suspected HIT have true HIT
Only ∼ 10% of patients investigated for HIT have a laboratory profile consistent with this diagnosis. At the McMaster Laboratory, when we tested over a 1-year period samples referred from our local (Hamilton, ON) hospitals, only ∼ 8% (25 of 310) tested SRA+.24 When we tested sera referred from elsewhere,7 a slightly higher frequency (188 of 1553 or 12%) was observed. In Europe, Greinacher et al found only 6% of 1650 referred sera were positive in a platelet activation assay (perhaps reflecting the greater use of low-molecular-weight heparin—and corresponding lower frequency of HIT—in Europe).25
Probability of HIT when the EIA is positive
Among EIA+ patients, the probability of SRA+ status (and thus, plausibly, HIT) ranges from 10%-50%, depending on the clinical setting. The lower value (10%) applies to low pretest probability settings, such as the ICU,4 or patients who present with VTE but without thrombocytopenia or other clinical features of HIT. For example, in the Matisse VTE trials, 127 patients tested EIA+ at study entry (presumably because of preceding heparin exposure), and only 14 (11%) also tested SRA+.3 Another setting in which EIA+ status confers low probability of SRA+ status is routine testing of postcardiac surgery patients: Pouplard et al21 found that only 11 (7%) of 163 EIA+ patients also tested SRA+.
The probability of a positive EIA indicating a positive SRA rises to ∼ 50% when HIT testing is performed because of clinically suspected HIT (data obtained from a mixture of clinical settings).22 This “50% rule” was also observed in a study of sera referred to a central laboratory for investigation of clinically suspected HIT reported by Greinacher et al: 95 (47%) of 204 EIA+ sera tested additionally positive for platelet-activating antibodies in the HIPA assay.25
In my practice, I do not usually use the EIA for diagnosis. The McMaster Platelet Immunology Laboratory performs the SRA twice weekly (Tuesdays and Thursdays), and I diagnose HIT based on the SRA results. We perform an IgG-specific EIA on Fridays, mainly as an additional “quality control” maneuver for the SRA. In other words, if the expected EIA+ status is not observed for an SRA+ result, one must consider whether the SRA was falsely positive (eg, due to thrombin contamination, presence of non-HIT platelet-activating antibodies, etc). In addition, we use the EIA-IgG as an independent arbiter for the minority of samples (∼ 2%-3%) that repeatedly produce platelet activation at all heparin concentrations (including at 100 IU/mL), a so-called SRA-“indeterminate” reaction profile.26 Of course, irrespective of the ultimate diagnosis based on the SRA result, my interim management is based on my clinical judgment on the likelihood of HIT (discussed subsequently in “My therapeutic approach to suspected HIT”).
DTIs versus indirect anti-Xa inhibitors
Three non-heparin anticoagulants, danaparoid, lepirudin, and argatroban, are approved for treatment of HIT, although approvals vary by jurisdiction (notably, danaparoid is neither approved nor available in the United States for the treatment of HIT).27 Three other agents, fondaparinux, desirudin, and bivalirudin, are marketed for non-HIT indications, but each has a biological rationale and some published data for therapy of HIT.14,28–34 These 6 drugs can be classified into 2 groups: those exhibiting long-acting, AT-dependent inhibition of factor Xa (danaparoid and fondaparinux) and the (AT-independent) DTIs (r-hirudin [lepirudin and desirudin], argatroban, and bivalirudin).
Table 2 summarizes key differences between these groups of drugs, emphasizing specific implications for HIT.30 As indicated by the check marks (✓), the long-acting Xa-inhibiting drugs have several advantages over DTIs.
A comparison of two classes of anticoagulant used to treat HIT
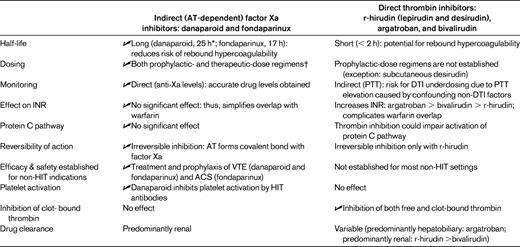
Check mark ( ) indicates favorable feature in comparison of drug classes. Reprinted, with modifications and permission, from Warkentin 2010.30
) indicates favorable feature in comparison of drug classes. Reprinted, with modifications and permission, from Warkentin 2010.30
ACS indicates acute coronary syndrome; INR, International Normalized Ratio.
*For danaparoid, half-lives of its anti-thrombin and its thrombin generation inhibition activities (2-4 h and 3-7 h, respectively) are shorter than for its anti-Xa activity (∼ 25 h).
†Although therapeutic dosing is recommended for HIT, availability of prophylactic-dose regimens increases flexibility when managing potential non-HIT situations.
Danaparoid, the only anticoagulant evaluated for HIT in a randomized controlled trial, became available in Canada in the early 1990s initially through a compassionate-release program and later as a Health Canada–approved treatment. In my experience, this agent has high efficacy and safety for treating HIT.35 Further, it is the only anticoagulant that inhibits HIT antibody–induced platelet activation through disruption of PF4/H complexes.36 I have used it successfully in patients with severe HIT: one notable patient exhibited microvascular limb ischemia and a platelet count nadir of 2 × 109/L; he made a full recovery (with limbs intact) on therapeutic-dose danaparoid.30 This patient also had overt (decompensated) disseminated intravascular coagulation, which is a risk factor for DTI failure.30,37
In February 2009, a global shortage of danaparoid meant that this drug became unavailable in Canada for 2 years. During this period, my colleagues and I used fondaparinux to treat HIT.34 We treated 16 patients with SRA+ HIT (mean, 91% serotonin-release; range, 68%-100%); 9 (56%) of the patients had HIT-associated thrombosis. No new thrombotic events occurred after starting fondaparinux, and uneventful transition to warfarin was readily accomplished. One patient who developed a calf hematoma was regarded as having a major bleed.
These data support the view emerging in the literature that fondaparinux is likely an effective treatment for HIT. Table 3 summarizes studies with a minimum of 5 patients each.31–34 The overall new thrombotic event rate was low (0% [0 of 52], upper 95% confidence interval, ∼ 7%) although a key caveat is that only the McMaster study reported on SRA+ patients. Moreover, experience with severe HIT (eg, platelet nadir < 20 × 109/L with consumptive coagulopathy) is minimal. The major bleeding rate (∼ 2%-5%) seems to be no worse than that seen with danaparoid (∼ 7% in pooled studies27 ) and DTIs (∼ 1% risk of major bleed per treatment day, or 8%-15% per treatment course27,38 ) when given in therapeutic doses to treat acute HIT. Long-acting anticoagulants may be problematic, however, in early postoperative or critically ill patients who are at high risk of bleeding.
Fondaparinux for treatment of acute HIT: studies including 5 or more patients with putative HIT (with or without HIT-associated thrombosis)
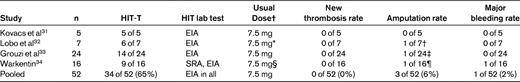
A PubMed search performed on April 23, 2011 using “fondaparinux” and “heparin-induced thrombocytopenia” had 152 “hits,” from which 3 studies31–33 of non-critically ill patients were identified that described the use of fondaparinux for 5 or more cases of acute HIT. Also included is one study by the author.34
*The single patient without thrombosis received 2.5 mg daily by subcutaneous injection.
†One patient with popliteal artery thrombosis and failed thrombectomy required amputation; however, irreversible ischemic limb necrosis was judged to have been present before fondaparinux therapy.
‡One patient had limb amputation before commencing fondaparinux.
§Dosing ranged from 2.5-15 mg/d (7.5 mg was the median daily dose).
¶One patient with brachial artery thrombosis and failed thrombectomy required amputation; however, irreversible ischemic limb necrosis was judged to have been present before fondaparinux therapy.
Although fondaparinux may itself rarely cause HIT when used for postoperative thromboprophylaxis,39 this possibility should not deter its use in treating HIT.28 The reason is that fondaparinux-associated “immunogenicity” (ie, its potential to trigger platelet-activating antibodies and, rarely, HIT) and its “cross-reactivity” (ie, its potential to promote activation of platelets in the presence of HIT antibodies, however triggered) are “dissociated” phenomena.40 When fondaparinux does trigger HIT, the pathogenic “autoimmune-like” antibodies activate platelets irrespective of whether fondaparinux or heparin is present.28 Indeed, fondaparinux is an unlikely potentiator of thrombocytopenia when given to a patient with HIT antibodies.3
Disadvantages of DTI therapy
Table 2 lists certain disadvantages of DTI therapy that are not seen with danaparoid and fondaparinux. For example, PTT monitoring of DTI therapy can be confounded by HIT-associated consumptive coagulopathy.30,37 In such patients, protocol-driven interruption of DTI therapy due to “supratherapeutic” PTTs, but not because of supratherapeutic drug levels, can be associated with progression of thrombosis (although this problem can be overcome by measuring DTI levels directly, these assays are hardly used in North America). In addition, DTIs have relatively high bleeding rates, which is of particular concern when so few patients with suspected HIT actually have this diagnosis.
My therapeutic approach to suspected HIT
When I evaluate thrombocytopenic patients for HIT, I ask the following questions:
1. What is the overall likelihood of HIT? For patients in whom the risk of HIT is judged low, options include continuing heparin (unfractionated or low-molecular-weight) or low-dose danaparoid or fondaparinux. (A further option might also be low-dose desirudin,29 although this is not currently available in Canada.) Especially if the patient is at risk of developing HIT over the next few days (eg, a postcardiac surgery patient I am evaluating for early postoperative thrombocytopenia), I often recommend prophylactic-dose danaparoid or fondaparinux—even when the patient clearly does not have HIT—rather than reintroduce heparin and potentially revisit the HIT issue again in a few days if the thrombocytopenia persists or worsens. For patients in whom the probability of HIT is considered high, I generally opt for therapeutic-dose danaparoid or fondaparinux regardless of whether thrombosis is present. When the probability of HIT is judged to be intermediate, I consider both prophylactic and therapeutic dosing as options, depending on relative perceived bleeding risk and HIT risk. When HIT is strongly suspected (or confirmed by SRA), I perform lower-limb ultrasound for deep-vein thrombosis evaluation and postpone warfarin (if required) pending substantial resolution of thrombocytopenia (platelet count > 150 × 109/L).27 (Warfarin use during acute HIT is a major risk factor for venous limb gangrene.27,30 )
2. Is there a definite non-HIT indication for therapeutic-dose anticoagulation? Although strong suspicion for HIT itself is a reason for therapeutic-dose alternative anticoagulation,27 there may be patients in whom the probability of HIT is judged to be only low or intermediate, but in whom there is nevertheless a strong (non-HIT) indication for therapeutic-dose anticoagulation, such as atrial fibrillation or acute thrombosis.
3. What is the patient's renal status? Both danaparoid and fondaparinux are renally excreted, and adjustments should be made for impaired renal status. For renally compromised patients who require therapeutic dosing, the initial danaparoid maintenance dose (after the usual bolus loading dose) is 25%-30% lower than usual (eg, ∼ 150 U/h rather than ∼ 200 U/h). For fondaparinux, the initial daily dose or 2 is as per usual (7.5 mg/d for a patient weighing between 50 and 100 kg), but then we use anti-Xa levels to judge subsequent doses; often, maintenance doses are only 2.5 or 5 mg daily.
4. Could the patient require an invasive procedure within the next few days? Both danaparoid and fondaparinux are long-acting anticoagulants (with half-lives of 17 and 25 hours, respectively, for anti-Xa activity) without antidotes, and thus caution should be used if a patient will undergo an invasive procedure. DTIs may have theoretical advantages in this situation, although one should keep in mind that in a patient with true HIT, interruption of short-acting DTI therapy can result in rapid recurrence of the prothrombotic state.
Final thoughts
The DTIs lepirudin and argatroban may be approved for HIT, but their true efficacies remain somewhat uncertain. Neither DTI was studied for HIT using randomized trials (rather, both were evaluated in prospective cohort studies using historical controls). The current American College of Chest Physicians–recommended lepirudin dosing protocol28 has not been prospectively studied, and the dosing regimen evaluated in the approval trials differs from the FDA-approved package insert. The argatroban trials did not require positive HIT testing, and many enrolled patients likely did not have HIT (whereas the historical controls were HIT test positive). Furthermore, as mentioned earlier, the safety and efficacy of the DTIs in non-HIT settings are largely unknown.
In contrast, danaparoid and (especially) fondaparinux have been shown to be safe and effective for preventing and treating thrombosis in diverse clinical settings. Moreover, major bleeding rates appear to be lower than for the DTIs. Emerging experience (Table 3) suggests that fondaparinux is also efficacious for treating HIT (although greater experience is required before its true efficacy for SRA+ HIT can be gauged). Given that ∼ 90% of patients evaluated for HIT ultimately are shown not to have this diagnosis, the option to use a long-acting, AT-dependent Xa inhibitor (in either prophylactic or therapeutic dosing as warranted) deserves consideration in the management of suspected HIT.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from GlaxoSmithKline and Gen-Probe GTI Diagnostics; has consulted for GlaxoSmithKline, Gen-Probe GTI Diagnostics, and Canyon Pharma; holds patents with or receives royalties from Informa; has been affiliated with the speakers' bureau for Pfizer Canada, Sanofi-Aventis, and Canyon Pharma; and has given medicolegal testimony relating to heparin-induced thrombocytopenia. Off-label drug use: Danaparoid and fondaparinux and bivalirudin and desirudin as “off-label” treatments for HIT.
Correspondence
Theodore E. Warkentin, Department of Pathology and Molecular Medicine and Department of Medicine, Michael G. DeGroote School of Medicine, McMaster University; Hamilton Regional Laboratory Medicine Program, Room 1-270B, 237 Barton Street East, Hamilton, ON L8L 2X2, Canada; Phone: (905) 527-0271 ext. 46139; Fax: (905) 577-1421; e-mail: twarken@mcmaster.ca.
