
Abstract
Advances in the treatment of myelodysplastic syndromes (MDSs) over the last decade have given patients and their hematologists a multitude of treatment options. Therapeutic options now exist that reduce disease-related symptoms, improve quality of life, and alter the natural history of the disease. Three drugs are now specifically Food and Drug Administration-approved for treatment of MDS: (1) azacitidine, (2) decitabine, and (3) lenalidomide. Clinical results with each of these agents, plus results with immunosuppressive therapy, are reviewed to guide clinical decision making. Although each therapy has made a substantial impact in improving the care of patients with MDS, unfortunately MDS treatment in 2010 ultimately fails in most patients, but these therapies provide a foundation on which we can build to further improve outcomes.
“The future ain't what it used to be”—Yogi Berra (Hall of Fame baseball player for the mid-century New York Yankees).
This is an odd way to begin a review on myelodysplastic syndromes (MDSs). It succinctly summarizes the main accomplishment of MDS research over the last decade: moving beyond the despair of the past and providing hope that advances can in fact be made. Here, I review clinical trials in MDS treatment, focusing first on clinical outcomes with new MDS therapies and then providing my own perspective on best practice guidelines for these treatments including optimal dosing, schedule of administration, and management of toxicities. Finally, progress in molecular understanding of MDS is discussed, particularly in the context of currently available therapies and a few “Yogi-isms” are included along the way.
Hypomethylating Agents: Results From Clinical Trials
Azacitidine
Azacitidine (AZA) was approved by the US Food and Drug Administration (FDA) on the basis of CALGB (Cancer and Leukemia Group B) 9221, a randomized trial comparing AZA with supportive care in 191 patients with MDS, targeting high intermediate and high-risk disease and including low-intermediate risk patients with ongoing complications of cytopenias.1 AZA was given at 75 mg/m2 subcutaneously for 7 days and repeated every 4 weeks. The results demonstrated superiority of AZA over best supportive care (BSC) in terms of quality of life, reduced transfusion needs, and delayed time to acute myeloid leukemia (AML) transformation or death for higher risk patients (21 months for AZA vs 12 months for BSC [P = .007]). Early transformation to AML (within 6 months) occurred in only 3% of patients versus 24% of those with BSC. Complete remission (CR) occurred in 10% of patients treated with AZA, and overall response (CR + partial remission + hematologic improvement [HI]) occurred in 47% by IWG (International Working Group) 2000 criteria.2,3 In part due to the crossover design, a survival advantage was not demonstrated. Yet, the trial brought definitive data to support evidence-based treatment of MDS patients with the endpoint of clinical response and improved quality of life, including the typical older patient with MDS more likely to experience adverse events.
A second randomized trial, AZA-001, was conducted to define whether an endpoint not of response but overall survival (OS) could be met with hypomethylation therapy. Patients had more advanced disease than the earlier CALGB 9221 trial, and, at entry, they were assigned by the treating physician to a conventional care regimen of either BSC, low-dose cytosine arabinoside (LDAC), or intensive induction chemotherapy. Then, the patient was randomized to either AZA or this preselected conventional regimen. AZA significantly improved OS, with median OS of 24 months vs 15 months in the conventional arm (P = .0001).4 At 2 years, 51% (95% CI, 42.1–58.8) of AZA-treated patients were alive, compared with only 26% (18.7–34.3) of conventionally treated patients. The CR rate in AZA-001 was 17%, with an overall response rate of 49%, similar to the CALGB trial.
The AZA-001 data become more convincing, perhaps, when looking at different subsets of patients treated. There was no particular “winner” that drove the beneficial response. Response benefit was distributed fairly evenly across various groups of patients. In virtually every patient characteristic category one can think to look at, there was a survival benefit to AZA over conventional care. This was true across younger and older patients, gender, performance status, FAB subtype, WHO (World Health Organization) classification (e.g., RAEB-1, RAEB-2, or other), International Prognostic Scoring System (IPSS) score, IPSS cytogenetic risk category, and IPSS bone marrow blasts. Similarly, survival and time to progression favored AZA, compared with each form of conventional treatment. For patients randomized to AZA versus BSC, median OS was 21.1 months versus 11.5 months, respectively (P = .0045). For patients randomized to AZA versus LDAC, survival was 24.5 months versus 15.3 months (P = .0006). For patients randomized to AZA versus intensive chemotherapy, however, the survival comparison did not show benefit to AZA, although there appeared to be a trend favoring AZA, 24.5 months versus 15.7 months (P = .51). The smaller sample size in this group limited statistical analysis.
The rate of CR was rather low in the CALGB and AZA-001 trials. The low CR rate provides compelling data against oncology dogma that achievement of CR is required for survival benefit. Indeed, a retrospective subset analysis examined the survival impact of AZA excluding patients who achieved CR.5 One-year survival rates were superior for AZA treatment versus conventional treatment, 68% versus 56% (P = .015), respectively. Thus, response less than CR was still associated with improved survival.
Decitabine
“Like deja vu all over again”—Yogi Berra
Decitabine is another azanucleoside that has followed a developmental path similar to AZA. Decitabine was approved by the FDA on the basis of a randomized phase III trial versus BSC using the European schedule of 15 mg/m2 intravenously over 3 hours, every 8 hours for 3 days, repeated every 6 weeks.6 Not embraced in the United States due to logistical issues and patient convenience, this schedule of decitabine showed a CR rate of 9% and an overall response rate of 30%. Decitabine-treated patients had improved quality of life and reduced transfusion needs. No survival benefit was shown in the trial, although there was delayed time to AML transformation or death higher risk disease, 12 months versus 6.8 months (P = .03). A more recent phase III randomized study (EORTC [European Organization for Research and Treatment of Cancer] 06011) compared decitabine with the European schedule versus BSC in 233 higher risk patients with MDS7 ; results have to date not been published. Abstracts reported that there was no OS benefit, although improved progression-free survival was noted in the decitabine arm. OS was a median of 10.1 months for decitabine versus 8.5 months for BSC (P = .38). The overall response rate for decitabine was 34%.
Limitations in trial design, from the perspective of year 2010 understanding of hypomethylating agents, may have negatively affected outcomes. One major limitation of the study was the relatively short duration of therapy administered. The median number of cycles per patient was four, and 40% received two or fewer cycles. By comparison, the median number of cycles of AZA given in AZA-001 was nine.4 The best available evidence, and consensus, is that patients benefiting from therapy with hypomethylating agents should continue treatment until progression or unacceptable toxicity. In a comparison across several different decitabine trials, patients treated for an extended duration appeared to have had better response.8 As such, it is possible that results from the negative randomized trial with decitabine might have been better if prolonged therapy had been administered. In addition, it is unclear whether the optimal decitabine schedule was used in the trial. In North America, the typical schedule is 20 mg/m2 intravenously over 1 hour daily for 5 days, repeated every 4 weeks, based on promising high CR results of up to 39% in a single center.9 A multicenter study of 99 patients with MDS using the same regimen, The Alternative Dosing for Outpatient Treatment (ADOPT) trial, showed a CR rate of 17%, with an overall response rate of 51% including HI.10 Whether using this schedule of decitabine for an extended duration would improve survival is unknown. Due to financial and logistical reasons (including patent expiration), as well as the now-defined superiority of AZA over conventional care, another trial of decitabine versus BSC in MDS will not be done. A comparative trial of AZA versus decitabine recently opened to accrual (http://www.ClinicalTrials.gov; identifier: NCT01011283). Unfortunately, the trial is designed with a primary endpoint of early response at 6 months and is not powered to examine the more relevant question of OS.
Strategies to Maximize Patient Benefit With Hypomethylating Agents in Clinical Practice
“You've got to be very careful if you don't know where you're going, because you might not get there”—Yogi Berra
Higher risk MDS patients who are of reasonable performance status with adequate organ function are excellent candidates for treatment. Lower risk patients who are transfusion dependent or have severe cytopenias are also appropriate candidates for hypomethylating agents, although goals of therapy and what constitutes “acceptable” toxicity may be somewhat different. Critical to the success of either AZA or decitabine is the application of repetitive cycles of therapy and, in turn, selection of appropriate patients who are willing and able to undergo prolonged therapy. Patient expectation of immediate disease response, and disappointment at the lack thereof, is likely an important contributor to the “treatment fatigue” that is a problem with any chronic therapy (especially one with meaningful toxicities such as AZA or decitabine). It negatively impacts on success. Thus, pretreatment counseling to patients should describe not only response and/or survival benefit, but also the time course of response. Understanding the potential treatment benefit of these agents without CR (the most likely scenario) is important. A patient once relayed to me the perfect analogy for explaining hypomethylation therapy, and I often use this story to educate other patients about the process. “MDS is like a car heading at high speed toward a brick wall; ideally, one would like to stop the car and get out. The next best thing is to slow down and enjoy the view.”
Azanucleosides: How Much? How Frequent? How Long?
In patients with higher risk disease treated for a survival endpoint, use of AZA 75 mg/m2 subcutaneously for 7 days, every 4 weeks, is most appropriate. Intravenous administration is also FDA-approved and has similar bioavailability.11 The 7-day approach every 4 weeks is feasible; in AZA-001, 86% of patients had no dose reduction, and 80% of cycles were given at 4- or 5-week intervals, without prophylactic myeloid growth factor use.4 Patients treated with AZA did not experience higher rates of infection or bleeding relative to the observation/conventional therapy groups (Table 1), a major finding given concerns of administering cytotoxic therapy to older patients with preexisting marrow dysfunction.2,12 Importantly, even the elderly group of patients older than 75 years had survival benefit (2-year OS of 55% [N = 38] vs 15% [N = 49]; P < .02) with good tolerability.13
Grade 3 or higher infectious toxicities with AZA in AZA-001 and CALGB 92221 compared with BSC, from Santini, et al (12)
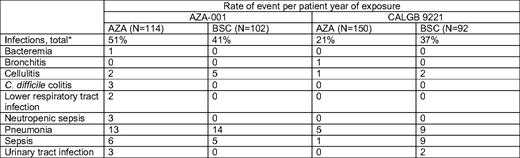
Includes patients preselected to BSC who subsequently received AZA.
*Includes other events not listed here.
Although alternative dosing schedules of AZA have been reported to have response rates that are similar to the published regimen from AZA-001, none of those schedules has been shown to improve survival.14 Use of 50 mg/m2 subcutaneously for 5 days every 4 weeks for patients with lower risk disease with a treatment goal of improved quality of life/reduced transfusions is reasonable if circumstances preclude administration of the 7-day regimen.14 A retrospective Spanish registry study of AZA in community practices provides evidence that fidelity to a 7-day schedule of AZA may be associated with better response.15 In 144 patients who received AZA for either 5 days, 5 days–weekend off–2 days (5/2/2), or seven consecutive days, those receiving seven total days of AZA had a higher CR rate, 22% versus 12%. Patients who received seven consecutive days had a higher overall response rate of 74%, compared with 65% for 5/2/2 or 58% for 5 days. Given the survival benefit and acceptable tolerability data from AZA-001, higher risk patients with MDS with AZA should be treated without reducing the dose or number of treatment days from the 75 mg/m2, 7-day schedule whenever possible.
For patients responding to AZA in AZA-001, the median number of cycles to best response was four; the median number of cycles to first response was two, and almost 90% of responding patients had response by cycle 6 (Table 2).16 Response tended to improve with continued treatment after first response, as 48% of patients had further improvement after initial response. Delayed response is not unusual; even for patients without objective response after six cycles (but with stable disease), 14% of patients subsequently had HI by cycle 9. HI was associated with a survival benefit.17 Thus, administration of six cycles of AZA before discontinuation, in the absence of intolerable toxicity or peripheral blood evidence of disease progression, should be part of the initial treatment plan. For patients without response (but with stable disease) after six cycles, continuation of treatment should be individualized. With decitabine, apparent improved efficacy and convenience suggest that 20 mg/m2 intravenously over 1 hour daily for 5 days every 4 weeks is the optimal regimen. In ADOPT, 68% of cycles were administered on schedule (if delayed, delay was a median of 8 days). Among patients in the ADOPT study who had benefit, 82% demonstrated first response by the end of cycle 2, and most responders also had best response at the same timepoint.10 I usually administer three to four cycles of decitabine without dose reduction before discontinuation due to lack of benefit. For both AZA and decitabine, maintenance of response with continued therapy is clearly important. Thus, in the absence of unacceptable toxicity or peripheral blood evidence of disease progression, therapy should be continued indefinitely for both. Whether younger patients with higher blast counts who have an immediate allogeneic transplantation option should receive hypomethylating agent therapy versus induction chemotherapy is unclear. No clear survival benefit was seen in the subset of patients randomized to intensive treatment in AZA-001, but decitabine appeared to have better outcome in high blast count patients compared with historical control in one retrospective analysis.18
Hypomethylating agents in MDS
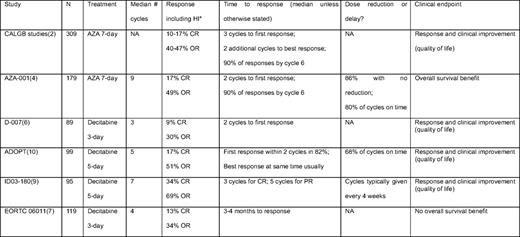
Table modified from Steensma and Stone, 2009.50
N, Number of patients treated with drug.
*Response by IWG 2000 criteria.
NA, data not available.
AZA 7-day; 75mg/m2 SC for 7 days, every 4 weeks; Decitabine 3-day; 15mg/m2 IV over 3–4 hours every 8 hours, every 6 weeks; Decitabine 5-day; 20mg/m2 IV over 1 hour for 5 days (and variants with total dose of 100mg/m2/cycle in ID03–180).
Adverse events are inevitably encountered in treating patients with MDS with these drugs. Hematologic toxicities are common to both hypomethylating agents and to the disease. Infections occurred in about 50% of AZA-treated patients in AZA-001 (vs 41% in the BSC group) and 58% to 61% of patients experienced grade 3/4 thrombocytopenia and/or neutropenia.12 Myelosuppression appears to be worse at least early on with decitabine than AZA, with slightly higher febrile neutropenia rates (10% hospitalized during cycle 1 in ADOPT10 ; 77% had grade 4 neutropenia and 28% had febrile neutropenia in the registration study6 ), but this problem is obviated by the likelihood that time to best response with decitabine is faster as well. Hematologic complications with both agents are worse in the first one to two cycles of therapy and are less frequent in subsequent cycles. These complications are, in my opinion, best managed with delay of the next cycle12 although a continued sense of urgency to give timely repetitive cycles is prudent to maximize benefit. In patients with neutropenia and infection during treatment, consideration of precycle neutrophil count is probably the best guide for decisions to delay therapy by 1 to 2 weeks, if necessary.12
Concern for infection in the setting of disease or drug-related neutropenia has understandably led to frequent prophylactic use of myeloid growth factors (granulocyte colony-stimulating factor [G-CSF]) in conjunction with hypomethylating agents in MDS. However, clinicians should be aware that there is currently little evidence that this improves clinical outcomes. Growth factors can add considerable cost and patient inconvenience to the regimen. By design, G-CSF was not used in the CALGB 9221 trial with AZA; it was permitted in AZA-001 in the setting of serious infection, but not given prophylactically. Yet, repetitive cycles in both trials were still given safely and in timely fashion without this prophylaxis, and infection risk was not substantially different from the BSC arm. Judicious use of G-CSF in the setting of ongoing neutropenic infection, particularly for patients with predominantly drug-associated neutropenia, is quite reasonable, but not necessarily mandated. I typically use G-CSF only in the setting of serious neutropenic infection when it is clear that the neutropenia is drug-related (eg, higher precycle absolute neutrophil count).
“When you come to a fork in the road, take it”—Yogi Berra
Impressive as the data are for hypomethylating agents in MDS, patients with higher risk disease should still be considered for frontline investigational therapy, although obviously the bar is higher now for success. Though the results with AZA and decitabine are academically exciting and a step forward, it is unlikely that any patient with higher risk MDS would be equally excited, given the prospect of only 50% survival in 2 years. It remains a sobering reminder that there is quite a bit of work ahead of us to improve outcomes further. Critical to further development of hypomethylating agent therapies are the identification of molecular/cytogenetic factors that may predict or enhance response. Included among these to date are the identification by our group of miR-29b as a possible positive predictive marker19 and apparent increased efficacy of the agents in patients harboring aberrations of chromosome 7.19–21 Microarray studies can predict phenotype and disease course in MDS22 and have shown that lack of response to hypomethylating agents may be related to overexpression of genes related to cell proliferation, providing new opportunities to understand and potentially overcome drug resistance in the future.23
Lenalidomide
Lenalidomide is an immunomodulatory drug that is FDA approved for lower risk MDS patients with transfusion-dependent anemia and an interstitial deletion of the long arm of chromosome 5 [del(5q)].24–26 It is active in a number of hematologic malignancies, yet its mechanisms of action remain incompletely understood. Numerous activities in different disease subsets include activation of cellular and innate immunity, enhancement of humoral antitumor immune response, inhibition of protein phosphatase 2A, induction of expression of the tumor suppressor SPARC, antiangiogenesis, and cytokine inhibition.27–32
Lenalidomide catapulted onto the international scene of MDS when the initial therapeutic endeavor (a single institution phase I/II trial) demonstrated remarkable efficacy in MDS with del(5q). In the initial study (MDS-001) of 43 red blood cell (RBC) transfusion-dependent patients with MDS (who were not neutropenic or thrombocytopenic), lenalidomide at 10 to 25 mg daily induced erythroid responses in 83% of patients with del(5q), compared with 57% of those with normal karyotype and 12% of those with other cytogenetic abnormalities (P = .007).33
Clinical Response With Lenalidomide in Lower Risk MDS With del(5q)
The subsequent trial of lenalidomide in lower risk, de novo MDS with del(5q) (MDS-003) enrolled 148 patients with the following additional eligibility criteria: RBC transfusion dependence (≥ 2 U/8 weeks), ANC > 500/μL, platelets > 50,000/μL, and low/Int-1 score by IPSS.34 Seventy-three percent of patients had failed prior erythropoietin treatment, 58% had received prior cytotoxic therapy. 74% had isolated del(5q), but only 27% had the classic 5q- minus syndrome described initially by van den Berghe.35,36 Treatment was lenalidomide, 10 mg daily, days 1–21 of 28-day cycles initially, subsequently modified for continuous dosing based on preliminary results of MDS-001. In the trial, 67% achieved transfusion independence (TI); 9% more had significant reduction in RBC transfusion needs. Time to response was about 1 month (median 4.6 weeks), and response was durable (62% of patients achieving TI remained transfusion-free for at least 1 year). Clonal elimination (complete cytogenetic response) occurred in 45% of patients, and 36% of patients had resolution of cytologic abnormalities.
To improve confidence in dosing efficacy and understanding of safety, a double-blind, placebo-controlled, phase III study examining two different doses of lenalidomide was performed in Europe for this same del(5q) patient population. Preliminary results of MDS-004 were reported at last year's annual meeting of the American Society of Hematology. In this trial, 51 patients received placebo, 46 received lenalidomide at 5 mg daily, and 41 received lenalidomide at 10 mg. In a modified intent-to-treat analysis, both lenalidomide arms were superior to placebo for TI and cytogenetic response. The rate of TI for each cohort was 61% for 10 mg, 50% for 5 mg, and 8% for placebo (P < .001, for each lenalidomide arm vs placebo), with 5.1 to 6.3 g/dL median increase in hemoglobin for lenalidomide responders. Complete cytogenetic response was 24%, 11%, and 0%, respectively (P < .001; P = .01 for each arm vs placebo).37 Median time to TI with lenalidomide was 3.3 to 4.3 months (range, 0.3–14.7).
Preliminary results of MDS-004 showed that lenalidomide treatment did not increase risk of AML progression, compared with placebo; this had been a significant concern in Europe, preventing approval of the drug. Rates of AML progression were 6% (5 mg), 1% (10 mg), and 2% (placebo), with median time to AML progression from the first dose of study drug of 9.3, 5.9, and 3.1 months, respectively. TI responses favored 10 mg as the recommended starting dose (as from MDS-003). However, nearly all patients required dose reduction or interruption due to myelosuppression. There was no apparent increase in hematologic toxicity for 10 mg versus 5 mg. These data support initiation of lenalidomide at 10 mg daily, despite a subsequent need for dose reduction in most patients.
Lower Risk MDS With del(5q) and Lenalidomide: How Much? How Frequent? How Long?
For those with del(5q) treated on MDS-003, dose adjustment from the starting dose of 10 mg daily was required in 84% of patients, including 91% of those on continuous daily dosing and 67% on 21-day dosing. At week 24 of the study, only 32% of patients were receiving 10 mg daily, 44% of patients were receiving 5 mg daily, and 24% of patients were receiving 5 mg every other day. The median interval to dose adjustment was about 3 weeks. Dose reductions were primarily due to myelosuppression; grade 3+ neutropenia occurred in 55% of patients; three patients died of neutropenic infection early in the treatment course. Grade 4 neutropenia was more common with daily dosing (44% vs 17%, P < .001), and 62% of grade 3+ hematologic toxicities occurred during the first 8 weeks of treatment. Thus, clinicians should follow blood counts weekly during the first 8 weeks of lenalidomide therapy. Creatinine clearance (CrCl) should also be followed, with dose adjustment if CrCl falls below 50 mL/min.
Interestingly, the development of cytopenias in del(5q) patients correlated with subsequent achievement of clinical response.38 Whether this toxicity is a prerequisite for response is uncertain, but the data suggest a direct cytotoxic effect of lenalidomide on the del(5q) clone. Several ongoing studies are examining the question of whether higher doses of lenalidomide (up to 50 mg daily), which cause increased myelosuppression, would increase the response rate in high-risk MDS and AML with or without del(5q).39 Prophylactic G-CSF is not necessary for safe administration of lenalidomide to del(5q) patients.40 In MDS-004, only 3% to 9% of patients received G-CSF prophylactically, although 17% to 26% of patients received G-CSF for treatment of infection/febrile neutropenia. For my clinic patients, I initiate lenalidomide at 10 mg daily, typically use a 21-day schedule of administration, repeat cycles every 4 weeks until progression, and hold/reduce the dose based on more than grade 3 neutropenia or thrombocytopenia, without G-CSF unless the myelotoxicity is prolonged.
Lenalidomide in Higher Risk MDS With del(5q)
A phase II study of lenalidomide at 10 mg daily was conducted in Int-2 and high-risk patients with MDS with del(5q).41 Other than the obvious differences in blast count (78% had blasts > 10%) and cytopenias and the adverse prognosis that goes along with those, another crucial difference in the population compared with that from the lower risk studies MDS-003 and MDS-004 is that few patients enrolled on the trial had isolated del(5q). In the higher risk trial, only 19% had isolated del(5q); 81% had del(5q) with one or more additional cytogenetic abnormalities. In contrast, in MDS-003 and MDS-004, approximately 75% of patients had isolated del(5q). This appeared to be a significant factor in response. In patients with higher risk MDS and one or more additional cytogenetic abnormalities beyond del(5q), only 1 of 38 patients (3%) had a response. However, in those with isolated del(5q), 6 of 9 achieved CR, with a median CR duration of 11.5 months; 4 of 6 patients with CR also achieved cytogenetic CR. Importantly, response occurred only in patients with presenting platelet count > 100,000/μL; 0 of 27 patients with lower platelet counts responded. Neutropenia (in those with baseline ANC > 1000/μL) was common, with grade 3+ in 79% with similar incidence of severe thrombocytopenia. Dose interruption and/or reduction were also common in this setting. I believe it is reasonable to consider lenalidomide as frontline treatment in the few higher risk patients with MDS who have both isolated del(5q) and platelet count > 100,000/μL, although it should be noted that this is based on experience in a very small number of patients.
Lenalidomide in Lower Risk MDS, Non-del(5q): How Much? How Frequent? How Long?
In a trial similar in design to MDS-003, except in the targeting of lower risk patients with MDS without del(5q) (N = 214), lenalidomide treatment led to TI in 26% of patients.42 Overall, 43% of patients had erythroid response, with time to response of about 1 month. Response duration was only 41 weeks, much shorter than in lower risk del(5q) patients. The median hemoglobin increase was 3.2 g/dL (about 40%–50% that seen in lower risk del(5q) patients). Of 47 patients with cytogenetic abnormalities at baseline, only four achieved cytogenetic CR. Study patients had ANC > 500/μL (only 27% of enrollees had ANC < 1500/μL) and PLT > 50,000/μL. Grade 3 or higher neutropenia or thrombocytopenia with lenalidomide was less common than in del(5q), occurring in 18% to 27% of patients. Continuous dosing versus 21-day dosing did not appreciably alter risk of myelosuppression. Dose adjustment was needed in 55% of patients, but the timing of this was much different than in del(5q). Instead of a median 3 weeks to this requirement, as with del(5q) patients, the median time-to-dose adjustment was 7 weeks. A follow-up double-blind, placebo-controlled, randomized study for non-del(5q) patients (MDS-005) to further investigate optimal dosing and toxicities, similar in design to MDS-004, is ongoing.
Lenalidomide and Molecular Biology
Molecular studies shed light on mechanisms of disease and lenalidomide's clinical activity in MDS. It appears that the drug may have different mechanisms of activity, depending on the disease type.26 In del(5q), lenalidomide suppresses the malignant clone, but in non-del(5q) it appears to promote erythropoiesis. Elegant studies by Ebert et al43 and others have examined genes in the 5q31-q35 region, ultimately implicating haploinsufficiency of RPS14 as the cause of MDS with del(5q), although this gene is apparently not the target of lenalidomide. 5q haploinsufficiency of two phosphatases important in cell cycle regulation, Cdc25C and PP2A, has been discovered; these phosphatases are the targets for lenalidomide.28 These phosphatases are directly or indirectly inhibited by lenalidomide, and this inhibition is a major factor in lenalidomide's clinical activity in del(5q) MDS patients.28 Finally, gene expression profiling can predict response to lenalidomide in patients with or without del(5q); patients with a defective erythroid gene signature are more likely to have an erythroid response to the drug.44
Immunosuppressive Therapy for MDS
Responses to immunosuppressive therapy (IST) of antithymocyte globulin (ATG) and cyclosporine (CsA) in MDS remain among the most durable (and perplexing) of all available therapies in the disease. Typically, equine ATG is given at 40 mg/kg intravenously for four consecutive days, in conjunction with methylprednisone and CsA. Initial reports of its use demonstrated achievement of TI in 21 of 61 (34%) of patients.45–47 More importantly, the probability of continued TI after 5 years was 76%. However, the toxicity of the regimen has led to considerable apprehension in its use. Most ATG studies in MDS have been single center experiences, and several reports in unselected patients have demonstrated lower response rates and significant toxicity with the regimen.48 Clearly, judicious patient selection for the therapy is critical for its success and productive implementation.
Helpful guidance for the use of ATG in MDS was recently published. Outcomes for patients with MDS who were given equine ATG (with or without CsA) in sequential protocols at the National Heart, Lung, and Blood Institute between 1971 and 2003 were described.49 For 129 ATG-treated patients, 39 patients (30%) responded; 9% had CR. Serious infusion-related toxicities were infrequent, but meaningful because 9% of patients required temporary intensive care unit support. Responses included 18 of 74 (24%) treated with ATG alone, 20 of 42 (48%) treated with ATG+CsA, and 1 of 13 (8%) treated with CsA. Median response duration was 3 years (3 months to 10 years), and median survival in the cohort was 10.5 years. Notably, factors affecting response were younger age (≤ 60 years), HLA-DR15 positivity, and use of combination ATG+CsA. There was no association of response with pretreatment marrow cellularity, paroxysmal nocturnal hemoglobinuria clone, or absolute neutrophil count. Furthermore, a multivariate analysis of the IST-treated group compared with results from a control group of 816 patients with MDS from the International Myelodysplasia Risk Analysis Workshop database showed that survival was improved in younger patients, those treated with IST, and those with low-intermediate IPSS risk scores.
The role of IST in MDS treatment remains enigmatic, and the serious toxicities that can be encountered with its infusion and long-term immunosuppressive effects have properly led to hesitation to its use in community and academic practice. However, in a manner similar to the rigorous selection of patients for transplantation, appropriate selection of patients for IST affords some the opportunity for prolonged responses without the requirement for repeated maintenance chemotherapy (such as AZA or decitabine). IST should be considered in lieu of a hypomethylating agent for previously untreated, younger patients with MDS with low or Int-1 risk disease with HLADR15, in accordance with the guidelines described previously.
Conclusions
“It ain't over til it's over”—Yogi Berra
The successes reviewed here are a result of decades of dedicated hard work in laboratory and clinical research, clinical expertise, and patient altruism, but they are only the beginning. Hopefully, the greatest academic benefit from each will be in providing momentum for the next decade of research. The next generation of advances will come from the discovery of new molecular targets and development of prognostic/ predictive markers for risk assessment and/or therapy selection, as well as development of novel therapeutic approaches, including allogeneic transplantation.
Acknowledgments
This work was supported by the National Institutes of Health/National Cancer Institute grants K23CA120708 and P50-CA140158.
Disclosures
Conflict-of-interest disclosure: The author receives research funding from Celgene.
Off-label drug use: None declared.
Correspondence
William Blum, MD, Division of Hematology and the Comprehensive Cancer Center, The Ohio State University, B310 Starling-Loving Hall, 320 W. 10th Ave., Columbus, OH 43210; Phone: (614) 293-3507; Fax: (614) 293-7526; E-mail: william.blum@osumc.edu
