
Abstract
Acute lymphoblastic leukemia (ALL) in adults is a rare disease. The results of therapy remain unsatisfactory, and progress has been relatively slow. This article will focus on curative therapy in patients aged 30 to 60 years, but will also discuss the management of elderly patients with ALL. Recent large trials have increased our knowledge of the factors that determine outcome, and have clarified the role of blood and marrow transplantation in the management of this disease. These trials have also highlighted the major issues we need to focus on if we are to improve outcomes. This article describes the results of chemotherapy and blood and marrow transplantation for Philadelphia chromosome negative and positive adult ALL in the “older” adult patient, but also critically examines the major controversies and suggests how they might be resolved. The role of allografting in adult ALL is comprehensively discussed. Results of recent studies on T-cell ALL and reduced-intensity allografting are reviewed. A better understanding of the biology of the disease (including gene profiling) may allow individualization of therapy and, in time, targeted therapy.
The long-term survival of “older” adults with acute lymphoblastic leukemia (ALL) who are intensively treated is about 40%1 (Figure 1). Hematologic remissions are obtained in over 90% of patients,1 and the depth of these remissions using flow cytometric and molecular techniques is the subject of current studies. It is likely that, with time, new response definitions based on these tests will be established. We divided adults into age < 30 years and 30 to 60 years because this seemed clinically relevant, and available data best dealt with these age categories. However, these divisions are not absolute or evidence-based, and an individual's biologic age and general fitness are of paramount importance. Some of the “pediatric” approaches being applied to younger adults may, in time, be applied to this group if current studies report positive findings.

There seems little value in directly comparing the outcome of adults with children in general because the disease has different biologic characteristics, and the tolerance to very intensive therapy is less. However, there are lessons to be learned from our pediatric colleagues, and many aspects of the therapy of children can be selectively adapted to adults. There are no randomized studies in older adults that demonstrate “pediatric” approaches to be superior and, indeed, the single-arm studies are still small scale in this age group, with insufficient follow-up. Much is unknown, but the wide variety of trials being conducted in adults with ALL is heartening (Table 1).
Selected studies in Philadelphia negative and Philadelphia positive ALL
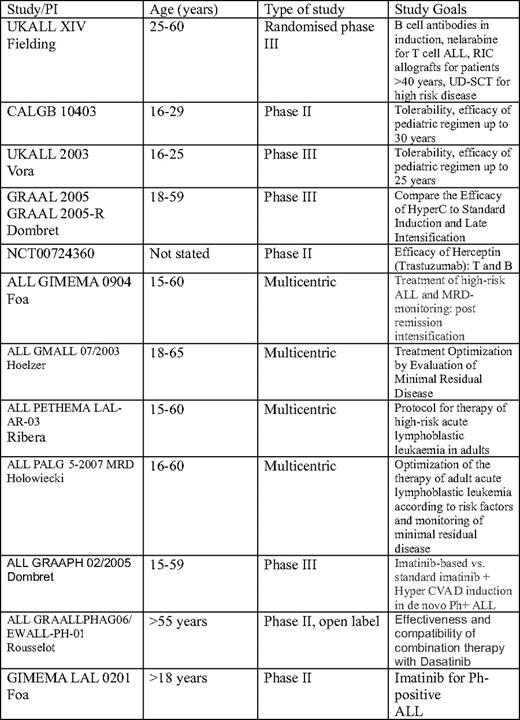
CALGB, Cancer and Leukemia Group B; GRAAL, Group for Research on Adult Acute Lymphocytic Leukemia; GIMEMA, Gruppo Italiano Malatti EMatologiche dell'Adulto; GMALL, German Multicenter ALL; UD, unrelated donor; PETHEMA, Programa para el Estudio de la Terapéutica en Hemopatía Maligna; LAL, Spanish abbreviation for ALL; PALG, Polish Acute Leukemia Group; GRAAPH, Group for Research in Adult Acute Lymphoblastic Leukemia; Ph+, Philadelphia positive; GRAALLPHAG, Group for research on Philadelphia positive ALL.
Prognostic Factors
We now have a better understanding of the factors that determine survival, but these will require reexamination as we introduce novel therapies. Cytogenetic findings—such as Philadelphia chromosome positivity, t(4;11), complex cytogenetic abnormalities (more than five chromosomal changes), and low hypodiploidy/near triploidy—result in inferior survival.2 Some of these changes are more common in older adults.
Other conventional factors—such as increasing age, high white blood cell count, and B-cell disease (rather than T-cell disease)—still hold true and predict higher failure rates with standard chemotherapy. However, many of these factors are also associated with a higher relapse rate after allografting, and it is not necessarily the case that bone marrow transplantation (BMT) is the solution for patients with adverse prognostic features. Combining these factors may allow individualization of therapy, a prospect not previously possible in this rare condition.
As well as undertreating patients with ALL with chemotherapy that is likely to fail,3 prognostic factors should be used to avoid overtreating better prognosis patients with allogeneic transplants that have a high upfront risk and may result in chronic graft-versus-host disease (GVHD), infertility, and secondary malignancy. Chemotherapy and transplant have complementary roles in ALL management, and a pragmatic approach is required to deliver the best outcomes. The role of BMT is likely to increase,4 especially with the promising results of reduced-intensity allografting,5 but conversely the use of BMT should be reduced if advances in nontransplant therapy improve cure rates.
Induction Strategies
The goal of remission induction therapy is to achieve remission without undue toxicity with a haematologic recovery that permits further therapy to be promptly given. Most regimens use prednisolone or dexamethasone, vincristine, daunorubicin, and asparaginase, with later exposure to cyclophosphamide and ara-C (cytosine arabinoside or cytarabine). HyperCVAD (cyclophosphamide, vincristine, doxorubicin [also known as adriamycin] and dexamethasone), which does not contain l-asparaginase, achieves high complete remission (CR) rates in newly diagnosed patients and is a reasonable alternative for induction therapy, but has not been shown to be superior to more traditional induction protocols.
Dexamethasone is preferred to prednisolone because of superior lymphocytotoxicity, better central nervous system (CNS) penetration and fewer thromboembolic events; these data are derived from pediatric studies.6 Poly(ethylene glycol)-asparaginase may be associated with more effective asparagine depletion and this in turn may lead to better outcomes.7 But this requires a randomized comparison. The safety and optimum dose of this drug require study in adults.
B Cell and Other Antibodies
As in other B-cell malignancies, monoclonal antibodies to CD20 or CD228 are being tested as adjuncts to chemotherapy in the hope that they will increase remission depth and improve survival without increasing hematologic toxicity. Sixty percent to 80% of B-cell ALL patients express these antigens at variable densities, but there is little evidence linking antigen expression to response. CD20 expression may be associated with a worse prognosis, so it is logical to investigate CD20 antibodies in randomized trials,9 and it may improve outcome.10 Alemtuzamab (anti-CD52) is being tested in adults, but may increase infection rates.11
Philadelphia+ ALL
One-quarter of all adults are Philadelphia+ chromosome, and the incidence increases with age. Until the results of recent studies in older patients became available, most patients with Philadelphia+ ALL were managed with intensive chemotherapy and a tyrosine kinase inhibitor (TKI).12 Imatinib has improved the CR rate in a number of trials to > 90% and makes more patients eligible for transplant (Table 2).13–18 Imatinib-resistant mutations are increasingly reported, and these should be sought in relapsed and refractory patients.
Imatinib studies in Philadelphia positive ALL
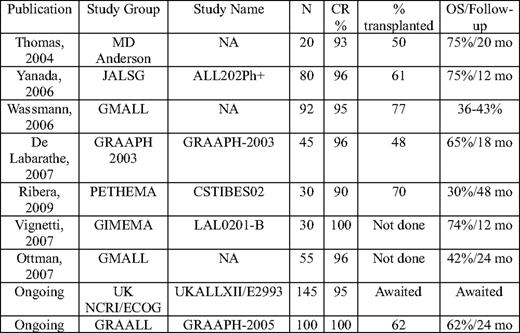
OS indicates overall survival; NA, not applicable; JALSG, Japanese Acute Leukemia Study Group; Ph+, Philadelphia positive; GMALL, German Multicenter ALL; GRAAPH, Group for Research in Adult Acute Lymphoblastic Leukemia; PETHEMA, Programa para el Estudio de la Terapéutica en Hemopatía Maligna; GIMEMA, Gruppo Italiano Malatti EMatologiche dell'Adulto; LAL, Spanish abbreviation for ALL; NCRI, National Cancer Research Institute; UKALL, United Kingdom ALL Group.
Adapted from a table supplied by Dr A.K. Fielding.
Dasatinib, which inhibits tyrosine and src kinases, holds considerable promise.19 It also may be effective in CNS disease.20 There are no randomized comparisons with imatinib, although it is a more potent inhibitor of tyrosine kinase in vitro. Recent studies from Italy and France with dasatinib alone in older patients have achieved very high remission rates with encouraging short-term survival. Good minimal residual disease (MRD) responses correlated with outcome. Data regarding the combination of dasatinib and intensive chemotherapy are lacking. It is possible that less conventional induction therapy may be required and that allogeneic stem cell transplantation (SCT) may not be mandatory.
The remarkable effectiveness of TKI therapy, in some studies without chemotherapy or allografting, has made us consider deescalation of therapy, but the long-term results of these less intensive approaches are unknown, and allografting is the only known cure. The effect of pretransplant MRD status on outcome is unclear.
A study of 267 patients (prior to the TKI era) showed allogeneic transplant to be superior to chemotherapy, with 44% and 36% surviving 5 years after sibling and unrelated donor SCT, respectively.21 However, only 28% of patients proceeded to a CR1 allograft, reducing its impact, and making it important that we improve nontransplant therapy (and improve access to transplant). The Minneapolis group reported 50% survival in 14 patients who received reduced-intensity conditioning (RIC) allografts from cord or sibling donors.22 TKIs were used only for morphologic or molecular relapse posttransplant. Studies of TKI posttransplant that examine dose, duration, and molecular response are urgently required; this is the subject of studies from the German and UK groups that are soon to be reported.
Consolidation, Intensification, and Maintenance Therapy
Little of this therapy is evidence-based in adults. The optimal number, intensity, and components of consolidation therapy are all uncertain. Many protocols use high-dose methotrexate and asparaginase, the former partly as CNS-directed therapy.
The value of maintenance has been shown by a recent randomized comparison of autograft and continuing maintenance showing the latter to produce 10% better survival.1
Supportive Care and Management of Complications
There is little published about this important area of ALL management, and a recent survey of the European Working Group on ALL (EWALL) members revealed wide variation in practice. These issues are particularly marked in “older” adults.
Infection: Patients are at high risk of bacterial infection during induction due to profound neutropenia and high-dose steroids. Many clinicians manage patients in an outpatient setting, but, occasionally, patients die of neutropenic sepsis. Ciprofloxacin or related agents are often given prophylactically, but the evidence that they reduce infection-related mortality is questionable, and they may increase the risk of Clostridium difficile. The incidence of probable and proven fungal infection is uncertain, but this is a not uncommon problem. Drugs such as itraconazole cannot be given concomitantly with vincristine (they increase bioavailability), and many use intermittent liposomal amphotericin or caspofungin. This requires prospective study and preferably a comparison of antifungal options, such as voriconazole or posaconazole, for which there is evidence of activity in acute myeloid leukemia.
Coagulation abnormalities during asparaginase therapy: Unfortunately, there is no clarity here, with many pediatricians advising against monitoring and only replacing clotting factors if there is bleeding. In adults, there is little evidence to guide practice, and most hemato-oncologists monitor clotting and use fresh frozen plasma and cryoprecipitate to normalize clotting (reviewed recently by Earl23 ). Thrombosis, it should be noted, is a more common problem.
Avascular necrosis: The crude incidence in one study was 4%, with a seemingly higher incidence in younger patients and, surprisingly, patients treated with chemotherapy.24 However, in my experience, allograft patients who receive total body irradiation (TBI) and steroids (usually for GVHD) have a high incidence of avascular necrosis.
Hepatotoxicity: About 10% of patients develop abnormal liver function tests, which are frequently drug-related. Cessation of drugs, such as proton pump inhibitors and cotrimoxazole, as well as antifungals (eg, ambisome or azoles) may result in improvement. Occasionally, the bilirubin becomes significantly elevated, and chemotherapy has to be delayed or dose-reduced, making these patients among the most problematic to manage.
T-Cell ALL
Last year, a UK–US collaborative group described the biologic and clinical features of 356 adults with T-cell ALL, as well as their therapy and outcome.25 This was the largest reported experience of homogeneously treated patients; several new findings potentially impact on decision making. The outcome is slightly better than for patients with B-cell disease, with 94% entering CR and 48% surviving 5 years. Blast expression of CD1a and lack of expression of CD13 were associated with higher survival (P = .01 and < .001, respectively), but with survival of about 60%, these subgroups are not candidates for deintensification of therapy. This study found evidence for a graft-versus-T cell leukemia effect with the donor versus no-donor analysis showing 15% superior survival if a matched sibling donor was identified. The predominant reason for better outcome was reduced relapse. The subgroups of patients with a white blood cell (WBC) > 100 × 109/L had poorer survival (P = .03), as did patients > 35 years, and those with a complex karyotype. Allogeneic transplant should be prospectively assessed in all poor prognostic groups to establish if it is better than chemotherapy. Nelarabine, which has promising results in patients with relapsed or refractory disease, also should be tested upfront in this group of patients.26 Finally, CNS disease (occurring in 9%) did not affect survival; these patients should be aggressively treated and are curable.27
CNS-Directed Therapy
CNS involvement occurs in 5% of adults at diagnosis and although it impacts on survival (29% vs 38%; P = .03), it is unclear how much prophylactic CNS-directed therapy is required.27 Most protocols do not now give CNS irradiation, and 4 to 6 intrathecal doses of chemotherapy and high-dose methotrexate seem to be adequate to prevent CNS relapse. For patients proceeding to TBI-containing allograft regimens, intrathecal chemotherapy alone is sufficient. More prophylaxis may be required with RIC regimens, but this remains untested. CNS involvement is more common with high WBCs, T-cell disease, and a mediastinal mass. For patients with CNS involvement, there is a suggestion that allografting may result in superior outcomes, but there are no randomized data confirming this.
Relapse
The goal of relapse therapy is to obtain a remission without undue toxicity and to move to allogeneic transplantation as rapidly as possible. Many patients die shortly after relapsing, and many do not achieve CR2. A study of 607 patients who relapsed and had a 5-year survival of 7% is a sanguine reminder of the current limitations of therapy.28 The rates of CR2 were not documented, but patients who were transplanted with sibling or unrelated donors experienced only 24% and 16% survival, respectively. Survival was slightly better in patients age < 20 years and those with a CR1 duration > 2 years. In a French study, 44% of relapsed patients achieved CR2, but only 12% survived.29 We should learn two things from these data. First, at present, our best therapy should be given upfront because we cannot rely on salvage if primary treatment fails. Second, to improve outcomes, there needs to be a focus on the management of relapse. There is no standard of care for relapse, but the author recommends FLAG (fludarabine + cytosine arabinoside + granulocyte colony-stimulating factor [G-CSF]) for patients with short remissions30 and a standard four- drug reinduction (or FLAG) if remission is > 2 years while carefully maintaining awareness of the potential cardiotoxicity of anthracyclines. An SWOG (Southwest Oncology Group) study of mitoxantrone and high-dose ara-C in relapsed and refractory ALL achieved remissions in 23% (7 of 31 patients), but no long-term survivors.31
Role of Allogeneic BMT
Matched Sibling Donors
A major unresolved controversy is the value of sibling allografting in CR1.32 Approximately one-quarter of patients will have this option. The recent UKALL (UK Acute Lymphoblastic Leukaemia) XII/ECOG (Eastern Cooperative Oncology Group) 02993 study (in which patients had sibling allografts, irrespective of risk status) showed that patients with sibling donors had an 8% higher chance of survival at 5 years (53% vs 45%), but this survival advantage did not hold for patients age > 40 years because of a high transplant-related mortality (TRM) (35% at 2 years).1 The logical next step is to investigate RIC allografting in this older age group and to compare it with chemotherapy in those without a donor. Donor versus no donor analyses, which are designed to avoid biases favoring those who are able to proceed to allograft, may underestimate the positive effect of allografting because not all patients with matched sibling donors receive the assigned therapy.
This trial justifies the use of myeloablative (full-intensity) sibling allografting in patients age < 40 years, but further studies are needed to refine this strategy. A subset of patients (eg, women wishing to retain fertility) may be able to avoid allografting, but this seems only justifiable in patients with no high-risk factors. For those with access to molecular MRD, a negative result after 10 to 16 weeks of therapy may provide some confidence in continuing with chemotherapy. It is also possible that patients age > 40 years who have no comorbidities may benefit from full-intensity allografts. If the TRM could be minimized, this group would benefit from considerable reduction in relapse risk that allografting provides.
Transplant in Specific Subgroups
Most allograft centers would perform an allograft in CR1 using a sibling or well-matched unrelated donor in t(4;11) disease, but outcomes are still poor.33 Many of these patients present with a high WBC count and do not achieve deep remissions prior to transplant. There is insufficient evidence to recommend a CR1 allograft for patients with the t(1;19), and not all studies have identified this as a high-risk cytogenetic abnormality with intensive therapy. The Center for International BMT Research (CIBMTR) compared TBI with either etoposide or cyclophosphamide. Provided > 13 Gy of TBI was used, the two agents were equivalent in CR1 patients, but etoposide/TBI was superior in CR2 patients.34
Unrelated Donor Transplantation
A recent report described encouraging results with allografting from unrelated donors (URDs) for patients with Philadelphia-negative ALL in CR1 at high risk of relapse.4 About 40% of patients, 90% of whom had high-risk features, survived 5 years. The following factors were associated with poor survival: WBC > 100 × 109/L, > 8 weeks to CR1, cytomegalovirus seropositivity, HLA mismatching, and T-cell depletion. Relapse risks were modest; TRM is the major cause of treatment failure. Selecting closely HLA-matched URDs and reducing TRM should improve results. Other studies have found that URDs and sibling donors yield similar results.35 A further study from the British Society of BMT using partial in vivo T-cell depletion with Campath antibodies showed excellent survival and a low incidence of GVHD.36 Age was not a significant prognostic factor in either study, but these findings should be applied with caution when applying URD SCT to patients age > 40 years.
Allogeneic Transplant in Second Remission
Allogeneic hematopoietic cell transplantation (HCT) is the treatment of choice for adults with ALL in CR2 with sibling allografts achieving disease-free survival of 25% to 40%. For other patients, URD SCT should be pursued, and for those without well-matched donors, cord blood or even haploidentical SCT at experienced centers should be performed because chemotherapy alone cures very few adults.37
Allografting for Patients Not in Remission
There are few data that support this practice, but a recent CIBMTR study showed that about 16% of patients can achieve 3-year survival with better results if the marrow blast count is < 20% and performance status is good.38 There is little place for allografting very unwell patients with refractory high blast count disease.
Reduced-Intensity Conditioning
Allogeneic BMT may be the most effective antileukemic therapy in adult ALL, but results are compromised by high TRM, approaching 35% in patients age > 35 years.1 In an attempt to avoid the high TRM seen with full-intensity HCT, but to exploit the graft-versus-ALL effect, investigators have been exploring the role of RIC regimens.
There are few published large-scale data about the efficacy of reduced-intensity allografting for older patients with ALL. The two largest series from the European Group for Blood and Marrow Transplantation (EBMT)39 and CIBMTR40 both indicate about 35% to 40% survival at 2 to 3 years, albeit in a selected group of patients.40 The outcome of patients not in remission was dismal. The paper by Mohty et al39 indicated better outcomes with chronic GVHD, whereas the CIBMTR series showed less relapse with acute GVHD, but not with chronic GVHD (P = .08).40 Only the EBMT series showed lower TRM with RIC (compared with full-intensity grafts); this may reflect the age and performance status of the patients transplanted. There was a suggestion of more relapse with RIC (relative risk 1.34; P = NS), and this requires verification in a larger patient group. A better outcome occurred with lower age, good performance status, CR1 disease, and sibling or well-matched URDs. The converse of this was a lower chance of success in patients with a poor performance status or a mismatched URD.
These preliminary, retrospective data do not provide sufficient basis for recommending RIC allografts in older or more infirm patients with ALL in CR1. However, RIC allografts are reasonable for older patients in CR2,40 if they are not fit for a full-intensity allograft. Large-scale prospective trials (such as UKALL XIV) are needed to confirm the efficacy of RIC allografting and to assess its optimum timing, conditioning agents, and target patient group. These trials are likely to provide further evidence of a graft-versus-leukemia effect and its ability to overcome the adverse prognostic impact of factors that predict failure of chemotherapy. Older patients (age > 40 years) tolerate intensive chemotherapy poorly and survival currently is poor; selective application of RIC allografting may improve their outcome.
Autologous BMT
Although this therapy still has its proponents the UKALL XII/ECOG 2993 study provided firm evidence that autografting in CR1 is inferior to continuous maintenance chemotherapy.1 An analysis of > 300 patients in three French trials also found no advantage for autografting.41 High-dose chemoradiotherapy and autologous stem cell rescue might be more effective in patients who are molecularly MRD negative, but there are no prospective data to support this contention. This may be an attractive option in older patients who tolerate maintenance chemotherapy poorly, but data are not available to support this practice. Similarly, postautograft maintenance therapy might mitigate the high relapse rate associated with autografting, but this approach seems to be exposing the patient to the worst of both worlds.
Patients Age > 60 Years
The median age at diagnosis of ALL is > 60 years; this is an important area of need, with few trial data to guide therapy (reviewed by Larson et al42 ). Important biologic differences include a reduced male:female ratio, more B-cell disease, and more coexpression of myeloid antigens. The proportion of patients with Philadelphia positivity may continue to rise with age, but this has recently been challenged in a population-based study (Moorman et al43 ). Older patients may have reduced renal function and appear to be more prone to mucositis with consequent effects on nutrition and mobility. Acute confusional states due to chemotherapy, infection, and metabolic disturbance may cause treatment delays. More elderly patients take regular medications, making drug interactions more likely; these drugs should be discontinued if possible.
There has been little improvement in survival in the last 20 years.44 Fewer patients are enrolled in prospective trials, and some received attenuated doses of myelosuppressive agents, making data interpretation difficult. Thirty percent to 70% achieve remission, but survival is brief. The chance of early death is as high as is chemorefractoriness.45 Vincristine, steroids, and l-asparaginase cause more toxicity in elderly patients, and anthracyclines may be hard to administer in those with impaired cardiac function. Liposomal vincristine and anthracycline are being tested in this age group in randomized trials (eg, The GRAALL-SA1 [Group for Research on Adult Acute Lymphocytic Leukemia] Study). Overall, there are few data to guide therapy, although EWALL have succeeded in treating elderly patients with very intensive protocols with better CR rates and reasonable short-term survival (data not shown). This is reasonable in a trial setting, but, in my view, can only be regarded as worthwhile if some patients are cured or significant numbers of patients have good quality survival outside the hospital. Older patients with Philadelphia– disease and significant comorbidities should be offered attenuated induction therapy with less steroid and G-CSF support because the evidence for the use of G-CSF is best in this group.46 Obtaining a CR and medium-term good quality survival are the goals. Autografting in MRD– patients should be investigated in these patients because it might save them 2 years of maintenance chemotherapy.47 Age-specific trials are required to improve outcomes in this challenging patient group.
Nearly all older patients with Philadelphia+ chromosome disease should be treated with a TKI, vincristine, steroid, and possibly reduced dose anthracycline. If they achieve CR, subsequent therapy should be individually tailored depending on toxicity, comorbidities, and possibly MRD status. B-cell antibodies also deserve to be tested formally in this age group; rituximab is well tolerated.
Future Developments
Although we should be pleased with recent progress, to move forward we should accept that, with “older” adults with ALL, we fail more often than we succeed and that recent progress has been slow. Some of this lack of progress is due to the rarity of the disease, and international collaborations will be necessary to answer the major remaining questions. Over 90% of patients enter remission; we now need to focus on the depth of that remission using MRD techniques.48–50 A better understanding of disease biology using existing clinical, cytogenetic, and immunophenotypic factors, together with genomic profiling51 may result in more rational and targeted therapy. Several groups are evaluating “pediatric”-inspired protocols in “older” adults, but it should be noted that some of these trials retain “adult options,” such as allografting for high-risk patients (other than Philadelphia+ ALL) and that the nonrelapse mortality of pediatric protocols has been high in patients age > 45 years.52 A degree of caution is required until more prospective data become available.
Hemato-oncologists are urged to enter patients into well-designed, prospective trials. Worldwide, there is inconsistency in management, and many patients are following trial protocols, but are “off-study” and their outcomes are not reported. Outcomes are modest, and patients deserve to have access to novel therapies that may improve their chance of cure; every adult with ALL represents an opportunity to improve our knowledge.
How I Treat: Philadelphia– ALL
A previously well 39-year-old woman presents with pre-B ALL, with a WBC of 50 × 109/L and normal cytogenetics. After entering her on an MRD feasibility study, which uses the UKALL XII protocol, she receives a standard four-drug induction regimen. She enters CR at day 28 and a marrow shows < 0.01% of cells bear the leukemic immunophenotype. After recovery from phase 2 induction, her marrow is molecularly MRD–(< 104), although there was only one informative genetic marker making the result inconclusive. She has one child, but wishes to explore therapy that might result in retained fertility. Her sister is HLA-matched. I discuss three treatment options with her: (1) continued chemotherapy, (2) full-intensity allografting with etoposide, and 13.2 Gy TBI (3) or a reduced-intensity allograft with fludarabine, melphalan, and alemtuzamab (30 mg on day -1). With continued chemotherapy, she has a 30% chance of relapse,48–50 but also some risk of nonrelapse mortality and loss of fertility. Although full-intensity allograft has a 20% to 25% chance of TRM and a significant chance of chronic GVHD, this is my advised course of action even though it will almost certainly result in loss of fertility. I cannot recommend RIC allografting at this point (in the absence of prospective trials), but this may offer a lower TRM and some chance of cure.
How I Treat: Philadelphia+ ALL in Older Patients
A 65-year-old man with a history significant for smoking, heavy alcohol intake, and angina presents with CD20+ Philadelphia+ ALL and a white cell count of 20 × 109/L. His ECG (electroencephalogram) shows lateral T-wave inversion, and his echocardiogram shows a left ventricular ejection fraction of 48%. His goal of therapy is to survive 1 year when his daughter is getting married. I recommend treatment with vincristine, prednisolone, four doses of rituximab and imatinib 600 mg/day. He has one dose of daunorubin (30 mg/m2), which results in reversible atrial fibrillation. He enters CR by morphology and cytogenetics at day 28 and requires only 3 days of hospitalization. We meet to discuss postremission therapy and decide to administer a 50% dose of cyclophosphamide, ara-C, and 6-mercaptopurine (as per the UKALL XII protocol) while continuing imatinib. He has one episode of hypotensive neutropenic sepsis, which responds briskly to fluids and antibiotics. We then shift to 6-mercaptopurine and imatinib (400 mg/day) maintenance long term. (Figure 2)

Adjusted probability of overall survival for patients in the reduced intensity (broken line) and full intensity conditioning groups of allogeneic transplants for ALL
Adjusted probability of overall survival for patients in the reduced intensity (broken line) and full intensity conditioning groups of allogeneic transplants for ALL
Acknowledgments
Adele Fielding critically reviewed the manuscript and kindly permitted access to unpublished work. Jenny Bird also reviewed the manuscript and made several helpful suggestions.
Disclosures
Conflict of interest disclosure: The author declares no competing financial interests.
Off-label drug use: None disclosed.
Correspondence
David I. Marks, MB, PhD, FRACP, FRCPath, Professor of Haematology and Stem Cell Transplantation, Adult BMT Unit, University Hospitals Bristol NHS Foundation Trust, Bristol BS2 8BJ, UK; Phone: +44 117 342 8523; Fax: +44 117 342 8628; e-mail: David.Marks@UHBristol.nhs.uk
