
Abstract
Chronic myeloid leukemia is a model of how the molecular understanding of a disease can provide the platform for therapy and diagnostics. Clinicians are now empowered with first- and second-generation tyrosine kinases, as well as molecular tools to monitor disease and characterize resistance. However, there are still unanswered questions regarding optimization of therapy, the utility of molecular monitoring, and the search (or need) of “cure” that bears thought. In this review, we will discuss these issues, as they provide a roadmap for what may lie ahead in the therapy of other hematologic malignancies, particular the other myeloproliferative syndromes, where specific genetic lesions, and targeted therapy, are now being realized.
Chronic myeloid leukemia (CML) is the “poster child” of translational medicine. The discovery of the Philadelphia chromosome,1 and the subsequent finding of the BCR-ABL chimeric gene, lead to an unique understanding of the biology of the disease that spurred the development of targeted therapy, as well as methods for the molecular monitoring of the disease.2 In combination, these pieces have been built into a therapeutic framework that is the envy of oncology.
In summary, here is where we stand in CML therapy: first, tyrosine kinase inhibitor (TKI) therapy is remarkably effective for chronic phase CML. The majority of patients (∼ 80%) who are treated in the chronic phase achieve a complete cytogenetic remission (CCyR) (Table 1); and those who achieve a CCyR have an excellent survival (∼ 90%).3,4 However, some patients become resistant and progress. The phenomenon of resistance increases with the stage of disease least in chronic phase, more common in accelerated, and virtually the rule in blast crisis.5,6 Relapse often stems from a point mutation in the Abl tyrosine kinase domain, which affects TKI binding and Abl inhibition.7,8 Imatinib resistance can be treated by “second-generation” TKIs (dasatinib or nilotinib), although CCyR can only be achieved in roughly 50% of patients with chronic phase patients with imatinib (IM) resistance, with less success in advanced-phase disease.9–13 Lastly, monitoring endpoints correlate with long-term outcomes, and guidelines have been established to judge response using cytogenetic, polymerase chain reaction (PCR), and mutation testing.
CCyR rates for approved tyrosine kinase inhibitors (TKIs)
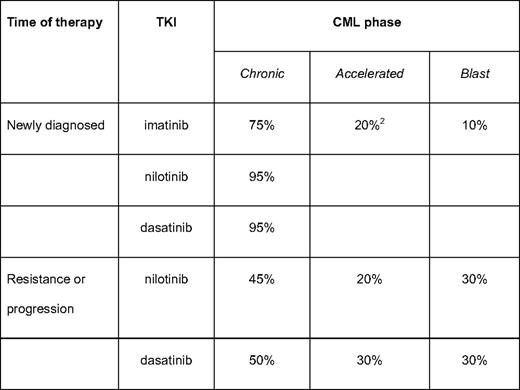
Estimates of CCyR, rounded to the nearest 5th interval for ease of memorization (3, 5, 6, 10, 12, 22, and 23).
The TKI era has dramatically affected CML. Age-adjusted US Surveillance Epidemiology and End Results (SEER) death data has shown a dramatic decrease in CML deaths, beginning with the introduction of imatinib (http://seer.cancer.gov/statistics/); no such change has been seen in other leukemia since the 1970s (Figure 1). However, even with this success, certain elements of CML therapy are posed for change. This article will discuss some of the cornerstones of CML treatment that seem likely to evolve in the near future, especially concentrating on features that are likely to prove relevant in the treatment of other myeloproliferative diseases.

SEER age-adjusted mortality rates. Rates are displayed for acute and chronic leukemia. The rates for CML are shown (purple triangles). The mortality rates in CML show a steady decline after the introduction of imatinib in the 1990s. By contrast, note the (humbling) lack of improvement in the other subtypes of leukemia.
SEER age-adjusted mortality rates. Rates are displayed for acute and chronic leukemia. The rates for CML are shown (purple triangles). The mortality rates in CML show a steady decline after the introduction of imatinib in the 1990s. By contrast, note the (humbling) lack of improvement in the other subtypes of leukemia.
Have We “Optimized” First-line Therapy?
Imatinib is highly effective in chronic phase CML.3 A 6-year follow-up of the pivotal phase III study in patients with chronic phase CML (IRIS [International Randomized Study of Interferon vs STI571]) (n = 553) showed rates of event-free survival (EFS) and freedom of progression to advanced disease of 83% and 93%, respectively (the 8-year follow-up continues to show a stability in response, with corresponding values of 82% and 92%; see Deininger et al13a ). Five-year follow-up and analysis of data from IRIS showed that patients who achieved a CCyR at 12 months were less likely to progress to advanced disease (progression-free rates: 97% vs 81%; P < .001).14 However, broader population studies show that resistance and intolerance to imatinib are more common than in the relatively controlled setting of clinical trials.15
The European Leukemia Net (ELN) and the National Comprehensive Cancer Network (NCCN) has provided remarkably similar recommendations for the treatment of CML.16,17 The ELN recommendations define an “optimal” cytogenetic response to first-line imatinib after 12 months of treatment as the achievement of a CCyR.14,18 Both the ELN and NCCN recommend that chronic phase patients begin on a “standard” imatinib dose of 400 mg/day. Patients who fail to achieve a complete hematologic response by 3 months, any cytogenetic response by 6 months, a major cytogenetic response by 12 months, or a CCyR within 18 months of the start of therapy are declared “failures” and should receive a second-line treatment.
How big of a problem is failure to imatinib? Actually, higher than one might expect at first glance of the excellent survival data. Using the ELN criteria, it appears that ∼ 25% of early chronic phase cases would be declared a “failure” or “suboptimal response” of imatinib.16,19 Another small proportion of cases will be intolerant to imatinib therapy, and thus also be candidates for secondary therapy. Now that we can define response criteria, and have alternative therapies, what should we do to prevent resistance?
The reflexive first approach is to just give more imatinib. The Tyrosine kinase inhibitor OPtimization and Selectivity Study (TOPS) compared imatinib 800 mg/day (n = 319) to 400 mg/day (n = 157). Higher dose imatinib provided faster responses CCyR rates at 6 months, 57% versus 45%, respectively; but, at 12 months, there was no significant difference between the arms (70% vs 66%, respectively).20 A 24-month update showed no significant difference between the cumulative CCyR rates (76% in both groups), EFS, progression-free survival (PFS), or overall survival.21 However, it is clear from these studies that those patients who can tolerate higher doses of imatinib, without interruptions for side effects, have a better response. Ongoing trials in both Germany (see Hehlman et al21a ) and France (see Guilhot et al21b ) combine imatinib with interferon and have demonstrated modest benefits in CCyR in patients receiving higher dose imatinib, as well as those with added interferon; it is not clear whether this will translate into long-term survival differences. It also is not clear how this strategy will compete with upfront therapy with second-generation TKIs.
Dasatinib and nilotinib are second-generation TKIs approved for the treatment of patients with CML resistant or intolerant to imatinib. Both agents are quite effective in resistant disease, yielding CCyR in approximately 50% of chronic phase cases (for cases who discontinue imatinib because of drug intolerance, the CCyR rates are > 70%). Because of this strong clinical activity, both drugs have been tried as first-line therapy in newly diagnosed chronic phase CML. Two single center trials of dasatinib and nilotinib have been performed at M.D. Anderson.22,23 At 18–24 months, both agents showed a similarly small, but consistent, benefit is CCyR over historical controls treated on imatinib trials. A phase II study from Italy of nilotinib in newly diagnosed chronic phase cases showed a similarly high rate of CCyR of > 90% after 12 months of therapy.24 Thus, there is much anticipation of the results of the industry-sponsored phase III randomized trials comparing standard-dose imatinib (400 mg) to either dasatinib or nilotinib. The preliminary data of the nilotinib data (the curiously acronymed “ENEST” trial; literary buffs ponder why Novartis could not find an “R” as homage to Hemingway) presented at the 2009 Annual Meeting of the American Society of Hematology (see Saglio et al24a ) suggest the following: (i) improvement of 12-month CCyR rates in the nilotinib arms, compared with imatinib (nilotinib 600 mg/day: 80% vs 65% for imatinib; nilotinib 800 mg/day: 78% vs 65%); (ii) better major molecular response (MMR) rates in nilotinib arms, compared with imatinib; and (iii) fewer progressions to advanced-phase disease in the nilotinib arm. Although these results look quite promising, the trial curiously found a substantially lower MMR rates in all arms, compared with those expected from historical controls. At the time of this writing, a few significant events are on the near horizon. First, both Novartis (nilotinib) and Bristol-Myers Squibb (dasatinib) will be facing off with the US Food and Drug Administration to gain approval for frontline chronic phase CML. Also, the preliminary results of the industry-sponsored phase III dasatinib trial should surface. Lastly, the results of the US intergroup trial comparing imatinib and dasatinib should be completed and reported. Thus, very soon, the strategy for the treatment of newly diagnosed CML may change. The obvious debating points are (i) whether or not a small benefit in early cytogenetic response with the second-generation TKIs will translate to a long-lasting survival benefit: (ii) the wisdom of switching to second-generation drugs without long-term safety data; and (iii) cost issues, especially with the prospect of generic imatinib in the foreseeable future.
Is Resistance Forever?
One of the unifying features of the biology of CML is the genetic instability caused by Bcr-Abl.25,26 Conceptually, both the processes of progression and resistance can be tied to genetic instability. Thus, the interval from the acquisition of Bcr-Abl and diagnosis allows unopposed Bcr-Abl signaling, and thus the genetic bedlam leading to changes facilitating or causing either resistance or progression (Figure 2). This can explain (i) the fact that resistance is lowest in early-phase chronic phase (time from diagnosis to treatment of under 1 year), higher in late chronic phase, and higher yet in accelerated phase and blast crisis; (ii) progression to advanced-phase CML is associated with (if not defined by) the consequences of instability (ie, DNA breaks causing new clonal cytogenetic events, point mutations, etc); (iii) Abl point mutations can be found in late-phase chronic and advanced-phase CML even prior to initiation of TKI therapy27,28 ; (iv) gene expression array experiments have found substantial overlap between the signatures of progression and resistance to TKIs.29–31 From these observations, one could speculate that patients who relapse with point mutations might have a natural history of more rapid progression than expected. Before second-generation TKIs, this indeed was observed.32,33 Also, one might expect that, at the time of resistance, selective pressure facilitates the outgrowth of multiple-resistant clonal populations. These patients could be treated with a second-generation TKI, but a new process of natural selection would be expected to take place, whereby the new selective pressure of dasatinib/nilotinib simple forces resistant clones to eventual outcompete sensitive ones (Figure 2).

A highly simplified model of the natural history of CML. (a) The CML leukemia stem cell (blue) regenerates, but also gives rise to leukemia progenitors. Presumably early in the disease, most are sensitive to TKI. With time and unopposed Bcr-Abl activity, cells arise with mutations and/or genetic features of progression. The latter eventually have a greater proliferative edge, and progression to blast crisis occurs. (b) If a TKI is initiated early in disease, before genetic instability creates clones more prone to resistance and progression, the natural history may be aborted. This may be a partial explanation of why some patients in complete molecular remission may be removed from TKI therapy. (c) Once resistance allows unopposed Bcr-Abl signaling, more resistant clones can emerge. Thus, the likelihood of eventual resistance to the second TKI is high, as is the probability of new mutations emerging. Figure not to scale.
A highly simplified model of the natural history of CML. (a) The CML leukemia stem cell (blue) regenerates, but also gives rise to leukemia progenitors. Presumably early in the disease, most are sensitive to TKI. With time and unopposed Bcr-Abl activity, cells arise with mutations and/or genetic features of progression. The latter eventually have a greater proliferative edge, and progression to blast crisis occurs. (b) If a TKI is initiated early in disease, before genetic instability creates clones more prone to resistance and progression, the natural history may be aborted. This may be a partial explanation of why some patients in complete molecular remission may be removed from TKI therapy. (c) Once resistance allows unopposed Bcr-Abl signaling, more resistant clones can emerge. Thus, the likelihood of eventual resistance to the second TKI is high, as is the probability of new mutations emerging. Figure not to scale.
In fact, several studies demonstrate this point. A key study by Hughes et al34 studied patients who were placed on nilotinib after failure on imatinib. Approximately one-half of cases failed to achieve a CCyR, and these patients were divided into three groups. The first group did not have mutations at the initiation of nilotinib and had no mutations at the time of failure. This group apparently had resistance independent of Abl mutation (and, thus, independent of Bcr-Abl signaling?). The second group had a mutation and relapsed with the same mutation. This is a curious group: Why should they initially respond and then lose that response? One suspects this group is also showing mechanisms of resistance that might be somewhat independent of Bcr-Abl signaling. The last group had a mutation, and at failure had a different Abl mutation. This is the most obvious example of clonal selection. As expected, these mutations appear to have a lower in vitro sensitivity to nilotinib. The same experience has been found in trials of dasatinib, as well, where patients who fail second-line therapy have evolved with new mutations.35 And, as would be expected, with sequential clonal selection, the proportion of patients carrying the T315I “gatekeeper” mutation, which is resistant to both nilotinib and dasatinib, increases.
What are the implications of these observations? The first and most obvious is that once a patient fails primary TKI therapy, one cannot blithely expect treatment with a second-generation drug to singularly save the day. As noted previously, second-generation TKI causes a CCyR in only 50% of cases, and if it is believed that CCyR is a legitimate predictor of long-term success, it means that one-half of our patients need something better. That something usually means allogeneic transplantation or a clinical trial. TKI therapy has delegated transplantation to “salvage” status; but, although it is associated with considerable potential toxicity, it also is certainly highly effective. Contemporary data from several groups show survival rates in chronic phase to be greater than 85%.36–38 Like all other therapy in CML, success greatly declines in advanced-phase disease, with survival in the accelerated and blast phases of approximately 40% and 20%, respectively. This underlines the importance of proceeding to transplant before progression occurs, and this means early HLA typing, institution of unrelated searches, etc, while instituting second-line TKI therapy. Two studies have suggested measures of response to guide second-line TKI therapy, allowing physicians to declare failure early and expeditiously proceed to transplant. These include achieving at least a minimal cytogenetic response after 3 months of therapy with a secondary TKI and a major cytogenetic response by 12 months.39,40 Thus, one strategy would be to start a donor search at the first sign of resistance. As the process of HLA typing and donor identification can easily take 3 to 6 months, patients who have an inadequate response at that time point can be efficiently moved to transplant.
The second implication of genetic instability is the rationale for more vigorous suppression of Bcr-Abl activity at the beginning of treatment. Presumably, a more complete suppression of Bcr-Abl could curtail genetic instability and thus delay or eliminate the biological processes of progression and the creation of competing clones (Figure 2). The early results of the ENEST trial, mentioned previously, that suggested less early progression in the nilotinib arms, provide some weight to this argument. The contrarian point of view is that there will always be a significant proportion of CML cases that had the disease for years before diagnosis; and, for these cases, the damage is done—add whatever TKI you will, clonal selection will still proceed per Darwin's expectations.
The third implication is that we need ways to predict response to therapy in both the upfront and resistance settings.
Can We Predict Response?
In the debate of what therapies should be offered in newly diagnosed chronic phase CML (imatinib, imatinib + interferon, dasatinib, or nilotinib), biomarkers that would predict response would seem quite useful. However, despite much effort, there are no clear winners yet.
Functional Studies.
TKIs are among the class of drugs potentially imported and exported by drug influx and efflux pumps, respectively. For example, imatinib can be imported by OCT-1, and exported by ABCB1 and ABCG2. Several in vitro studies measuring OCT-1 activity by imatinib uptake in mononuclear cells have demonstrated that cells demonstrating high OCT-1 activity, compared with low activity, have higher response rates (in survival, EFS, molecular response, and mutation rate), compared with cells with lower OCT-1 activity.41–43 These data reiterate the fundamental importance of Bcr-Abl inhibition in response, genetic instability, and resistance. Other studies have suggested that mRNA OCT-1 levels (which are much easier to measure) also correlate well with response.44 However, although intracellular imatinib levels are influenced by OCT-1, the second-generation TKIs, dasatinib and nilotinib, are not. However for the later two agents, activity with ABC efflux pumps may be involved in maintaining intracellular drug levels.
Genetic Studies.
Single nucleotide polymorphism arrays can measure associations of specific gene polymorphisms with phenotype (in this case, response to TKI), whereas mRNA expression arrays can measure gene expression and pathways associated with response. The most direct evidence of polymorphism on response comes from the study of the ABCB1 (MDR1) polymorphisms. In a study of 90 CML patients, those with a homozygous allele 1236 TT polymorphism had higher plasma imatinib levels and a higher frequency of molecular responses; the authors did not demonstrate effect on intracellular imatinib levels.45 As noted previously, several studies have shown an overlap of the gene signatures of response and progression.29,31 Moreover, a recent paper suggested that the level of six reporter genes could clearly distinguish chronic from advanced-phase disease, and these same reporters faithfully distinguished newly diagnosed chronic phase from resistant chronic phase (which “looked” like blast-phase disease).30
Early BCR-ABL Response.
Perhaps the best predictor of long-term response is short-term response. The Australian group have clean and clear data (per usual), demonstrating that BCR-ABL PCR response after only 3 months of therapy is strongly associated with a variety of relevant treatment outcomes (achievement of CCyR, MMR, and better PFS).46 This is very important because it implies that, simply by gauging a short-term response, a long-term plan can be reliably devised.
Mutation Testing.
Testing for the ABL mutations is not useful in newly diagnosed chronic phase patients (though it should be done in advanced-phase cases, where mutations are commonplace). Both ELN and NCCN guidelines suggest mutation testing at the time of increasing Bcr-Abl levels, or at loss of response. The rationale for this approach is to first attempt to identify patients unlucky enough to have a T315I mutation. Because this mutation is not responsive to either nilotinib or dasatinib, patients who have lost response with this mutation should either go onto transplant or research trial. Second, it is suggested that such mutation testing might be useful if the patient has a mutation that is sensitive to one second-generation TKI, but not to the other. Does this make sense?
A recent Technology Assessment of the US Department of Health and Human Services Assessment of Healthcare Research and Quality (AHRQ) asserts NO! The AHRQ examined the > 3000 reports on mutations in CML and distilled that to 16 reports it felt had sufficient purity of essence to provide interpretable data (http://www.cms.gov/DeterminationProcess/downloads/id76TA.pdf). The AHRQ assessment concluded that, although T315I mutation testing was important, there was no clear data that testing of other mutations mattered, because the outcomes of patients with any mutation versus those without mutations were similar.
Admittedly there are some problems with mutation testing, that include the following: (i) how the in vitro sensitivity of mutations are performed and calculated; (ii) as methods of detecting mutations vary, and have different sensitivities, the significance of mutation detection may potentially vary from study to study; (iii) selection of patients entering studies, as well as definition of outcomes; and (iv) follow-up is not standard across studies. Nonetheless, some common clinical sense should be put to use (nicely reviewed by Branford et al47 ). The most glaring flaw in the AHRQ analysis is the reduction of mutations to a dichotomous variable (mutation present or absent), when the whole reason for testing is distinguishing between types of mutations. For example, it is true that, if one compares imatinib-resistant cases receiving nilotinib therapy, the CCyR for those with a mutation was 32%, compared with 40% without a mutation.16 However, the clinical utility of mutation testing is evident when the mutations are evaluated by those mutations with good versus poor sensitivity to nilotinib: a CCyR of 40% for good versus 0% for the poor sensitivity E255K/V, F359C/V, and Y253H mutations. The same trend is found in dasatinib trials of imatinib resistance, in which the CCyR of patients with mutations versus no mutations were 44% versus 56%, respectively); while of patients with mutations, those with sensitive mutations had a CCyR of 53% versus 32% with poor sensitivity mutations.35 Of note is that the nilotinib poor-risk E255K/V, F359C/V, and Y253H mutations appear sensitive to dasatinib; and the poor-risk dasatinib mutation F317L (which had a CCyR rate of 7% on dasatinib therapy) and the infrequent V299L appear sensitive to nilotinib. It is estimated that ∼ 40% of imatinib-resistant patients harboring mutations will have one of the mutations in which detection may influence decision making (T315I, E255K/V, F359C/V, Y253H, or F317L). Because roughly half of imatinib-resistant cases will have a mutation at the time of resistance, nearly one-quarter of cases will have mutation results that should be part of the decision process. To be sure, other factors (eg, potential toxicity profile of the second-generation TKI), as well as physician experience with the particular TKIs are also important to the decision-making process, but data suggest that knowing the mutation status of the patient is a common-sense ingredient of patient care and planning.
What Do We Mean When We Talk About “Cure”?
What does it mean to cure a patient with CML? According to the Oxford English Dictionary, it is “verb: 1 relieve (someone) of the symptoms of a disease or condition. 2 end (a disease, condition, or problem) by treatment or appropriate action. 3 preserve (meat, fish, etc.) by salting, drying, or smoking.” Definitions 1 and 2 are relevant to how we think about CML (thankfully, not definition 3). One routinely hears arguments that TKI therapy is not “curative,” as opposed to transplantation, which supposedly is “curative.” Is this a meaningful discussion? Is it even accurate? Is our goal to relieve symptoms or to end disease?
First let us consider patients treated with TKI therapy. For those who respond with a CCyR, late events of loss or response are rare. Some patients become negative by PCR, whereas most remain positive. In fact, some patients are able to come off therapy, and roughly 40% will remain in molecular remission after withdrawing TKI therapy (see Mahon et al47a ). In contrast, following allogeneic transplantation, roughly 10% of cases will be BCR-ABL+ by PCR after 5 years of follow-up, and most will not relapse.48–50 The mechanism for this apparent “dormancy” is unclear. Even after transplants, long-term relapses occur, at a rate of approximately 1% per year. The Center for International Blood and Marrow Transplant Research has reported on 2444 cases of chronic phase CML who survived 5 years after an allogeneic transplant.51 The 15-year overall survival for these 5-year survivors was 88% and 87% for related and unrelated transplants, respectively, with relapse rates of 8% and 2%, respectively. Thus, long-term relapses do occur, and, in this, the longest relapse took place 18 years posttransplant. At the Fred Hutchinson Cancer Research Center, 800 cases of CML are alive after transplantation for CML (T.G., unpublished data, 2010). Of these, 25 patients have relapsed at > 10 years of follow-up, including a twin at 25 years!
What does this mean in how we approach CML? Many studies have shown that the most primitive CML cell is resistant to TKI inhibition; intuitively, this is the potential reserve for clones available for resistance and/or progression. The early and late relapse data after transplantation suggest that these remarkable hardy CML “stem cells” are furthermore resistant to myeloablative therapy, as well as immunologic surveillance and attack. The fact that complete molecular response is relatively uncommon in TKI therapy, and common following an allograft, suggests that the allograft is more potent than TKI, but only to a degree. Given that it is likely mathematically improbable that all leukemia stem cells can be destroyed, perhaps we should use “cure” as a shorthand for relieving symptoms without detectable disease, freeing patients and doctors from the impossible expectation of eliminating all disease, forever.
Conclusions—What Else to Do?
Despite the remarkable progress in CML, we are posed to ask and answer some significant questions that will make an impact not only on the management of CML, but also perhaps shape how the study of myeloproliferative diseases progress. A “short list” of things on the research agenda includes the following: (i) we need to determine to best use of second-generation TKIs. If the state of the world economy and health costs, studies on how best to use a generic imatinib would seem prudent. Do we treat upfront with imatinib, then switch to more active agents if aggressive milestones are not reached? Or alternatively, give more potent second-generation drugs first and then switch optimal responders to maintenance therapy with a cheap imatinib? (ii) We need to conduct trials on how reacting to specific Abl mutations makes a difference, as well as determine if treatment of a molecular relapse (as opposed to cytogenetics) is useful. (iii) We need to determine if complete molecular response is a relative and useful endpoint. It is promising that ∼ 40% of patients in continued complete molecular remission who have their imatinib discontinued stay in complete molecular remission. Although it is reassuring that those who have relapsed have responded to the reintroduction of imatinib, one wonders what happened during those months of uninterrupted Bcr-Abl signaling? Did mutations arise leading to eventual resistance or progression? Detailed studies of these patients could prove very instructive. (iv) Lastly, we await effective therapy of the T315I mutation, lest it be the final destination of all resistance.
The next years promise to be fruitful, interesting, and, most importantly, helpful to our patients.
Acknowledgments
It is a privilege to be asked to write this education summary for the American Society of Hematology. I do so with a bit of reserve and a lot of humility, for there are many others who are far more accomplished and deserving. Hopefully, I was able to suitably honor their hard work in this manuscript. If there are any oversights or blunders, please forgive me. I also apologize to those readers who would have preferred a more global look at CML, as well as those who would have more enjoyed a focused, detailed review.
Disclosures
Conflict-of-interest disclosure: The author has consulted for Ariad, Axid, Pfizer, BMS, and Novartis; has received research funding from Novartis; and has been a member of the Board of Directors/advisory committees for Natavis.
Off-label drug use: None disclosed.
Correspondence
Jerald P. Radich, MD, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue North, D4-100, PO Box 19024, Seattle, WA 98109-1024; Phone: (206) 667-4118; Fax: (206) 667-2917; e-mail: jradich@fhcrc.org
