
Abstract
Clinical management of myelofibrosis (MF)—whether primary or arising from an antecedent myeloproliferative neoplasm (post-essential thrombocythemia/polycythemia vera MF)—is currently in a period of transition that began with the discovery of the JAK2-V617F mutation 5 years ago. Selective JAK2 inhibitors have been developed, and clinical trials thus far have demonstrated that several of these agents meaningfully reduce MF-associated splenomegaly and constitutional symptoms. JAK2 inhibitors have durable benefits, act across the spectrum of MF subtypes, and provide a level of symptomatic benefit not seen with previous generations of nontargeted therapies. However, the JAK2 inhibitors can cause anemia and/or gastrointestinal disturbance, and their impact on JAK2 allele burden and the natural history is not yet fully defined. Several additional therapies that do not directly target JAK2 (eg, immunomodulatory drugs, histone deacetylase inhibitors, and inhibitors of the mammalian target of rapamycin [mTOR]) may ameliorate MF-associated anemia and morbidity-inducing symptoms. Balancing the potential benefits of these new agents against the risks and benefits of allogeneic stem cell transplantation (which can be curative, but carries a high risk of treatment-associated morbidity and mortality) requires an accurate estimation of the prognosis for an individual patient. Enhanced prognostic modeling systems are helping us to better characterize prognosis in MF patients not only at diagnosis, but also along the dynamic and variable course of the illness. Future advancements in the efficacy of MF-targeted therapy will likely arise from new pathogenetic insights and from combining JAK2 inhibitors with other agents.
Myelofibrosis (MF) in 2010 remains the myeloproliferative neoplasm (MPN) that induces the most morbidity and is associated with the poorest life expectancy.1 Although pathogenetic origins may vary, clinically MF is an amalgam of individuals with apparently de novo MF (so-called primary MF or PMF), as well those who evolved from a clear antecedent MPN, either polycythemia vera (PV) or essential thrombocythemia (ET) (post-ET/PV MF).2 Regardless of subtype of MF, patients suffer from a spectrum of complications arising from the malignant clone, including ineffective hematopoiesis with resulting cytopenias, splenomegaly, bothersome constitutional symptoms, risk of vascular events (including thrombosis and hemorrhage), and risk of blastic transformation.3
Historically, management of MF has been divided between allogeneic stem cell transplantation for a small subset of young and severely afflicted patients, or modestly effective medications and other interventions designed to palliate either anemia (eg, androgens, erythropoietin, or thalidomide) or splenomegaly (eg, hydroxyurea, splenectomy, or splenic radiotherapy).4 In 2010, no US Food and Drug Administration (FDA)-approved medication carries a specific MF indication, nor has any agent clearly demonstrated an ability to alter the natural history of the disease.
The year 2005 heralded a watershed moment for MF with the discovery of constitutively activating mutations in the JAK2 tyrosine kinase, most commonly JAK2-V617F.5 JAK2 somatic mutations are present in about half of PMF and post-ET MF patients, and almost all post-PV MF patients. Subsequently, investigators observed several other clonally restricted MPN-associated mutations such as MPL-W515L/K6 and allelically heterogeneous mutations of TET2.7 The discovery of these mutations prompted rapid development of selective JAK2 inhibitors, with the first of these agents entering clinical phase I testing a little over 2 years from the discovery of JAK2-V617F (Figure 1). Rapid and complete remissions were anticipated with JAK2 inhibitors based on the tremendous success of targeted tyrosine kinase inhibition (TKI) with imatinib mesylate8 and subsequent TKIs in the related MPN of chronic phase chronic myeloid leukemia (CML). However, the reality of JAK2 inhibition in MF has been less exciting than imatinib in CML. Targeted therapy in MF is associated with some benefits, but responses are almost always incomplete, such that the optimal clinical management of an individual MF patient remains a complex matter. In an era of rapidly evolving options for MF patients, I believe that the challenges facing clinicians can be summarized by four key questions.

Question 1: How Can I Assess Prognosis and Risk of Complications in Patients With MF?
Patients with MF have a varied phenotype in terms of age and disease severity, and thus can have a diverse prognosis. An accurate assessment of prognosis is essential in choosing the appropriate therapy for an individual patient, given that some patients may live more than 15 years,9 whereas others, such as those who progress to acute leukemia, have a life expectancy as short as 2 months.10 Because the intensity of intervention may be as minimalist as periodic observation of blood counts without specific therapy or as aggressive as allogeneic stem cell transplantation, an accurate and ongoing assessment of risk is crucial.
Historically, peripheral blood findings such as at the time of diagnosis (eg, anemia) and changes in leukocyte count have been the most prognostically significant variables in MF, and were incorporated into the 1996 Lille criteria (Dupriez score)11 (Table 1). In 2009, the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) retrospectively analyzed over 1000 cases of PMF at diagnosis and developed a four-tier International Prognostic Scoring System (IPSS) incorporating five factors with independent negative prognostic impact: (1) age > 65 years, (2) anemia, (3) constitutional symptoms, (4) leukocytosis, and (5) circulating blood blasts (Table 1).3 Of these five factors, anemia was the variable with the greatest negative prognostic significance.3 Further efforts to apply the IPSS factors at any point in time along the course of the illness resulted in a dynamic IPSS (DIPSS),12 which demonstrated that the acquisition of these factors at any point along the course of the illness is detrimental, particularly anemia (Table 1). Additionally, within each IPSS category (low, intermediate-1, intermediate-2, and high risk), karyotypic analysis further stratifies prognosis,13 with sole abnormalities of chromosomes 9, 13, and 20 (ie, trisomy9, del(20), and del(13)) being favorable, and complex karyotypes (more than three abnormalities) and trisomy 8 being unfavorable. Finally, young patients (age < 50 years) can live in excess of 15 years, if they lack thrombocytopenia and have no more than one adverse IPSS risk factor.9
Prognostic scoring systems for primary myelofibrosis (PMF)
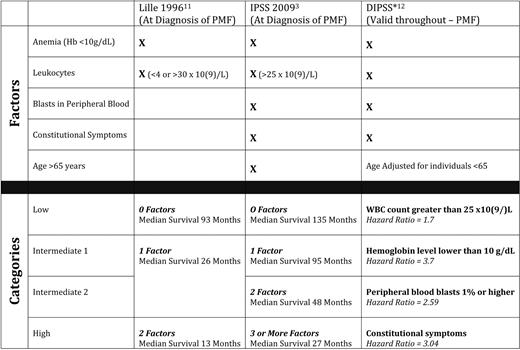
Hb indicates hemoglobin; WBC, white blood cell.
*Hazard ratio for the development of the risk factor during the course of MF in patients age < 65 years.
Current prognostication systems have several limitations. All current systems are derived from retrospective experience and solely from PMF patients (excluding post-ET and post-PV MF), and all systems incorporate clinicopathological variables that offer limited insight into mechanisms of disease progression, because these mechanisms are incompletely understood. Future insights into MF molecular pathobiology will hopefully allow us greater ability to predict disease progression and subsequent risk of complications or death from disease.
Question 2: Which Patients With MF Should Receive an Allogeneic Stem Cell Transplant in 2010?
Allogeneic stem cell transplant (ASCT) offers the possibility of cure to patients with MF.15,38–40 However, the risk of graft-versus-host disease (GVHD), transplant-related mortality (TRM), and posttransplant relapse of the MF itself make it essential to use this form of therapy only in the most appropriate candidates.
So, who are the optimal candidates for ASCT in MF? This is a highly controversial question. We have no randomized prospective data from which to draw conclusions. As with all decisions in medicine, the choice and timing of ASCT in MF is based on weighing of the risk-to-benefit ratio, as well as a thorough understanding by the patient of the complexity and potential adverse effects of the therapy they might choose to undertake.
Analysis of recent large series of ASCT in MF is sobering (Table 2), with TRM ranging from 10% to 30% (depending on age, donor compatibility, and conditioning regimen) and rates of acute GVHD (grades II-IV) ranging from 10% to 60%.15,38–40 Rates of chronic GVHD are even higher, with certain series reporting 85% of patients with grades II to IV chronic GVHD. Overall survival ranges from 30% to 60%; the largest retrospective International Bone Marrow Transplant Registry series suggests about one-third of patients with MF who are transplanted might expect long-term disease-free survival.14
Recent major series of allogeneic stem cell transplant (ASCT) trials for myelofibrosis
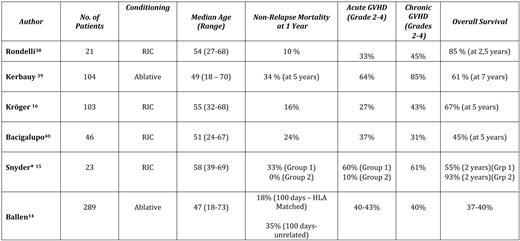
*Group 1, GVHD prophylaxis cyclosporine A + mycophenolate moffetil ± methotrexate; Group 2, GVHD prophylaxis tacrolimus/sirolimus ± methotrexate.
There are many factors that may potentially impact transplant outcomes, including choice of conditioning regimen, choice of GVHD prophylaxis, selection of donors, and selection of recipients. Unfortunately, given the overall rarity of MF, there are no randomized disease-specific trials comparing any of these variables in a structured manner. In general, it is believed, based on current series, that a reduced-intensity conditioning (RIC) approach is best and that use of umbilical cord stem cells is associated with a higher rate of graft failure; no regimens have been directly compared. Better results have recently been reported in patients submitted to RIC transplantation in a prospective way.16
Given the risks and unanswered questions in 2010, which patients with MF should be transplanted? The group of patients in which the risk-benefit ratio appears most favorable are those who are progressing toward blastic transformation. Given the median survival of patients after transformation is < 3 months,10 ASCT, if it is at all an option, should occur before transformation occurs. Indeed, the development of an “accelerated” phase of disease17 with increasing blasts (> 10%), worsening thrombocytopenia (< 50 × 109/L), and emergence of chromosome 17 aberrations are strong indications for ASCT in appropriate candidates, as determined by age and comorbidity status.
In the absence of increasing blasts, the IPSS would suggest individuals diagnosed with MF at an intermediate-2 and high-risk level would all have an expected median survival of 4 years or less. Such patients are potential candidates for ASCT, particularly if they have adverse cytogenetics.13 However, caution with younger patients (age < 60) is necessary, given that 3-year survival rates of younger patients in the intermediate- and high-risk IPSS groups are still 55% to 78% without stem cell transplantation.18 Patients in whom ASCT is feasible, but deferred at the time of diagnosis, should be followed; subsequent development of multiple adverse prognostic features from the DIPSS (particularly anemia)12 or an increasingly complex karyotype10 should prompt reconsideration of ASCT.
Question 3: What Is the Impact of JAK2 Inhibitors on Patients With MF?
Prior to the development of imatinib mesylate, ASCT was the only highly effective therapy for patients with chronic phase CML. The emergence of targeted TKIs dramatically decreased the necessity for ASCT in chronic phase CML. Has targeted medical therapy for MF similarly changed the timing of ASCT? In an era in which multiple novel agents are being tested for MF, how does their availability alter the management of current patients?
The last 3 years have seen an explosion of clinical trials for MF for two reasons. The first is that discovery of MPN-associated mutations (particularly JAK-V617F) identified plausible biological targets that small molecules could be developed to inhibit. The second reason is that the interest generated by scientific breakthroughs in MF and experience with CML suggested to the pharmaceutical industry that agents for MPNs could be profitable. Increased numbers of clinical trials in MF prompted development of standardized international criteria for biological response (IWG-MRT),19 with stringent criteria for complete response, partial response, and clinical improvement of anemia or splenomegaly. A need to quantify symptomatic burden (ie, fatigue, night sweats, fevers, weight loss, pruritus, and overall quality of life), which had previously been shown to be troublesome for patients with MF,20 led to the development of the Myelofibrosis Symptom Assessment Form (MF-SAF).21
Novel agents currently being tested in MF can best be broken down into two categories: (1) compounds used because of their ability to inhibit JAK2 (although off-target effects might account for some of the clinical effects), and (2) a group of diverse compounds aiming at targets other than JAK2 (Figure 1).
JAK2 Inhibitors
Inhibitors of JAK2 were first used in MF in 2007. All such agents available at present are molecules that target the native JAK2 and lack specificity for the mutant JAK2) (Table 3). The drug furthest along in the development process is INCB018424 (Incyte, Wilmington, DE), which has undergone trials in the United States and Europe). Results from a phase II study of 155 patients (a median of over 1 year on study) demonstrated that 50% achieved an IWG-MRT clinical improvement response for splenomegaly.22 In addition, serial administration of the MF-SAF throughout this trial demonstrated significant improvement in MF-associated symptoms.23 The greatest improvements in MF-SAF scores were reported by patients experiencing abdominal discomfort, night sweats, pruritus, and fever. Responses were equivalent regardless of subtype of MF (ie, PMF vs post-ET or post-PV MF) or JAK2 mutation status, and responses appear durable (median duration of response > 1 year). Although JAK2 was the intended biological target, decreases in JAK2 mutant allele burden were modest. The main adverse events were treatment-emergent anemia and thrombocytopenia, probably mediated by inhibition of JAK2 in native hematopoiesis. There can also be potential “rebound” effects with abrupt discontinuation of INCB18424 in MF, including recurrence of symptoms. Currently, two large phase III trials are ongoing in the United States and Europe, seeking regulatory agency approval of INCB18424 for MF.
Targeted therapeutic agents with reported effects 2010
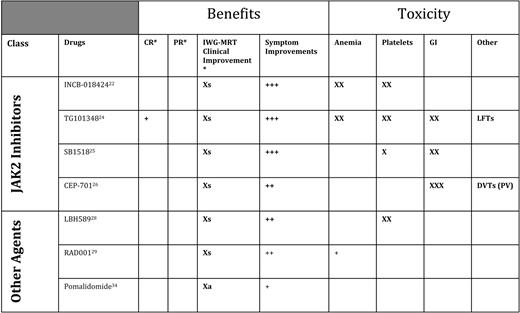
CR indicates complete response; PR, partial response; GI, gastrointestinal; Xs, clinical improvement for splenomegaly (> 50% reduction in palpable component if > 10 cm below left costal margin); LFTs, liver function tests; DVTs, deep vein thrombosis; Xa, clinical improvement for anemia (> 2 g/dL improvement durable > 2 months, or becoming transfusion independent).
*IWG-MRT responses.
Several additional JAK2 inhibitors are in earlier phases of testing, including TG10134824 (Targegen, CA), which appears to have similar activity INCB18424 with respect to improving splenomegaly and symptoms, but may (anecdotally) achieve slightly more significant reductions in JA2 allele burden and improve histologic features of disease. Adverse events of TG101348, in addition to anemia, include gastrointestinal disturbances. In a similar early phase of development is SB1518, a selective JAK2 inhibitor from S*Bio (Singapore); phase I/II trials in the United States and Australia have shown this agent to be similarly active for MF-associated splenomegaly and symptom burden.25 SB1518 has a potential advantage that thus far it does not seem to cause myelosuppression, but does cause gastrointestinal disturbances in some patients. Phase II testing is also proceeding with a new formulation of the JAK2 (and FLT3) inhibitor CEP-701 (Cephalon, Frazer, PA).26 In trials being conducted through MPD Research Consortium (New York, NY), CEP-701 has been associated with improvements in splenomegaly and symptoms, with gastrointestinal toxicity a limiting factor.27 A new formulation seems to be better tolerated and is promising. Other JAK2 inhibitors are in phase I testing, including CYT-387 (Cytopia, Melbourne, Australia) and LY2784544 (Lilly, Indianapolis, IN).
Additional Targeted Therapies for MF
Given that the JAK2 inhibitors have been active and helpful, but have only rarely induced complete remissions, therapies with other targets continue to be tested (Figure 1, Table 3). Trials of the histone deacetylase inhibitor LBH589 (panobinostat) from Novartis (Basel, Switzerland) in MF have demonstrated an ability to improve anemia and splenomegaly, and the agent is currently being tested in a phase II multicenter trial.28 Intriguingly, phase I testing results from Italy of everolimus (formerly RAD-001, an mTOR [mammalian target of rapamycin] inhibitor) (Novartis, Basel, Switzerland) have shown downstream inhibition of phosphoinositide 3 kinase, as well as indirect inhibition of JAK2; clinically, everolimus has induced improvement in splenomegaly and symptoms29 and is undergoing further testing for MF.
Immunomodulatory drugs (IMiDs) have improved cytopenias in patients with MF, beginning with thalidomide (either alone or in combination with steroids).30 However, use of thalidomide in MF is limited by neuropathy. Lenalidomide31 (alone or with steroids) has shown similar benefits (greatest in those with a del(5q)32 ), but its use is limited by excessive myelosuppression. In a large randomized phase II trial,33 pomalidomide (formerly CC-4047, Celgene Corporation, Summit, NJ) proved to be a well-tolerated agent for MF-associated anemia; even low doses of this agent (0.5 mg) are sufficient for reversing erythrocyte transfusion dependence in many patients.34 A randomized phase III trial of pomalidomide for improving MF associated anemia is planned.
Although significant therapeutic advancements have been made in the last 3½ years since the discovery of the JAK2 mutation, none of the novel agents have yet demonstrated a clear ability to induce durable complete remissions in MF, alter the natural history of disease, or prolong survival. Nor have we seen that medical therapy advancements in MF have reached the point where these newer agents would alter the decision on the timing, or appropriateness, of stem cell transplantation. How do we then build on the successes thus far observed in MF?
Question 4: How Can We Get Closer to Achieving Complete Remissions in MF?
In CML, it has clearly been demonstrated that, although TKIs may not eradicate the mutant clone, their ability to induce a complete hematologic, cytogenetic, and molecular remission allows control of the disease, arresting progression toward the accelerated or blast phase, and improves disease-free and overall survival when compared with prior therapies (eg, interferon and cytarabine).35 The reasons why targeted therapies have not been as effective in MF as in CML are numerous, not the least of which is the pathogenesis of the disease is clearly dependent on more than one pathway (ie, MF stem cells are not as “addicted” to mutant JAK2 as CML cells are to BCR-ABL),7 and MPNs that share a common mutation (such as JAK2-V617F) clearly have distinct phenotypes and natural histories. Further insight into the mechanisms not only of disease initiation, but also of disease progression are likely to be required to achieve the level of control in MF currently enjoyed by medical therapy in CML.
Until these breakthroughs occur, how might we make further incremental steps toward better outcomes in patients with MF? The current management of MF begins with an assessment of prognosis and typically has the clinician choose a single pathway of therapy, whereas in the future, hopefully, an accurate (and ongoing) assessment of evolving prognosis will be used to optimize combinations of therapies for the benefit of MF patients (Figure 2).

Currently, the experimental medical approaches in MF have focused (with the exception of occasional combinations of therapy with corticosteroids) on single-agent trials. Few malignant disorders have been successfully cured by single-agent therapy. Indeed, the birth of medical oncology as a field can be most accurately traced to the use of combination chemotherapy for the eradication of Hodgkin disease. What combinations might prove fruitful in MF?
ASCT remains the only curative therapy in MF, but with TRM, graft failure, and risk of relapse remaining. The JAK2 inhibitors have all shown varying degrees of ability to improve symptoms, performance status, and spleen size. There may be great benefit from combining a JAK2 inhibitor with ASCT to improve a patient's ability to tolerate an ASCT and reduce the spleen size to perhaps improve time to engraftment; a JAK2 inhibitor might also be used posttransplant to reduce risk of relapse. Indeed, the ability to decrease the spleen medically before transplant may be particularly beneficial because recent reports have suggested that, although prior surgical splenectomy may slightly improve time to platelet recovery after stem cell infusion, splenectomy prior to ASCT may impact GVHD effects and lead to a higher risk of relapse.16
The current spectrum of agents being tested for MF largely fall into two categories: (1) agents that lead to symptomatic relief with improvement in splenomegaly (with some either causing anemia or at least not improving anemia) or (2) agents that mainly improve anemia. A logical and greatly anticipated combination of therapies will be the combination of an IMiD to improve anemia (ie, pomalidomide) and a JAK2 inhibitor. The results are unpredictable. It is possible that there might be additive benefit, but there is also a risk that inhibition of the native JAK2 might inhibit normal hematopoiesis and thereby abrogate the beneficial effects of pomalidomide on anemia. Agents with alternative mechanisms of action (eg, everolimus, LBH589, etc) may also benefit from being used in combination with IMiDs, JAK2 inhibitors, or both.
The therapy of the earlier MPNs, ET, and PV may well be affected by the beneficial stem cell effects of pegylated interferon-α2a (peg-INFa) and might delay progression of these patients to post-ET/PV MF.36 If the process of MPN progression to MF were to be identified, these therapies could be combined in sequential fashion with peg-INFa in a comprehensive strategy to further suppress the disease. Indeed, even in CML, the combination of peg-INFa and TKIs is increasingly showing promise both for remitting and maintenance therapy.37 Finally, clinical trials with agents targeting mechanisms of fibrosing are planned (eg, with the TGF-β inhibitor GC-1008; Genzyme Corporation, Cambridge, MA).
The understanding and therapy of MF have advanced significantly over the past 5 years, with several approaches and agents making a meaningful difference for MF patients even today. Several trials seeking FDA approval for agents in MF are ongoing—a situation without precedent in this disease. Current management of our MF patients requires treating clinicians to assess prognosis accurately, consider risks and benefits of each line of therapy, and engage in a carefully shared decision-making process with the patient. Future advancements in therapy will likely result from combination therapeutic approaches, targeted therapies based on new pathogenetic insights, or both. Continued careful study of all of these options, and avoiding the temptation to quickly discard partially active therapies, are necessary for progress to continue.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from SBio, Novartis, Celgene, Incyte, Roche, Eisai, and Telik.
Off-label drug use: Various off-label uses in MF will be discussed—thalidomide, lenalidomide, azacitidine, and decitabine. There is no FDA-approved agent for myelofibrosis.
Correspondence
Ruben A. Mesa, MD, Professor of Medicine, Mayo Clinic, 13400 E. Shea Blvd., Scottsdale, AZ 85259; Phone: (480) 301-8335; Fax: (480) 301-4675; e-mail: mesa.ruben@mayo.edu
